FOR SUPPORT, CALL 1-866-APF-3635 (273-3635)
These Emergency Room Guidelines (download PDF) cover essential information for the emergency physician treating a patient in an acute porphyria attack, including common precipitating factors, typical presentation and other diagnostic clues, making the initial diagnosis, common sequelae and best practices for treatment. A PowerPoint presentation (as PDF) for instruction is also available.
Please note: These Guidelines are for Physician Use Only. The APF sells a separate, personalized Emergency Room and Primary Care Physician Kit that contains all the information acute porphyria patients need to have with them in the Emergency Room (medical journal articles, information about diagnostic labs and Panhematin®, room for your own diagnostic test results).
Key Points
Introduction
Clinical Features
Diagnostic Recommendations
Treatment
Bibliography
Neville R Pimstone MD, PhD, Head Liver Diseases Greater West Los Angeles Veterans Affairs, Professor Emeritus UC Davis
Karl E. Anderson MD, Professor, Departments of Preventive Medicine and Community Health, Internal Medicine, Pharmacology and Toxicology; Associate Director, General Clinical Research Center, Director, Porphyria Center and Laboratory, University of Texas Medical Branch, Galveston, Texas
Bradley Freilich, MD, Kansas City Gastroenterology and Hepatology, LLC
Porphyrias are inherited and/or acquired disorders of heme biosynthesis. A review article published in the March 2005 Annals of Internal Medicine (1) provides expert recommendations on the diagnosis and treatment of the acute porphyrias, which are the most life-threatening of the porphyrias. In addition, two valuable websites are available as resources for the ER physician:
American Porphyria Foundation (APF): www.porphyriafoundation.com
European Porphyria Initiative: www.porphyria-europe.com
Heme is synthesized in all tissues in eight enzymatic steps, and the end product is a component of many essential hemoproteins, such as hemoglobin, myoglobin and cytochromes, including the cytochrome P450 enzymes (CYPs) that are especially abundant in the liver. The first enzyme, ALA synthase (ALAS), controls the rate of heme synthesis in the liver. This enzyme is down-regulated by heme. The four acute hepatic porphyrias are due to different enzyme deficiencies (Table 1). The most common of these is Acute Intermittent Porphyria (AIP), but the others can produce the same neurovisceral symptoms. The enzyme deficiency limits the capacity of the liver to increase heme synthesis. Therefore, when drugs, hormones or other factors that induce ALAS and CYPs are given, ALA and porphobilinogen (PBG) are overproduced and accumulate, and a neurovisceral attack may develop (2). Both ALA and porphobilinogen (PBG) increase during attacks of AIP, Hereditary Coproporphyria (HCP) and Variegate Porphyria (VP), although these increases may be less and more transient in HCP and VP. Increases in porphobilinogen (PBG) are generally greater than ALA. In ADP, which is extremely rare, only ALA is increased. Increases in urinary porphyrins also occur in these conditions, but are less specific because they also increase in many other medical conditions.
Gastrointestinal symptoms:
Acute abdominal pain occurs in about 85-90% of attacks (1), and is neurologic in origin. The pain is usually severe, diffuse, unremitting for hours and poorly localized, but is sometimes colicky. Nausea, vomiting and constipation are common, but diarrhea is sometimes noted.
Neurologic symptoms:
Pain in the extremities, back, chest or head is also common. Objective sensory loss may be found in up to 40% of cases (1). Peripheral motor neuropathy is an indication of a severe and potentially life-threatening attack. Neuropathy can progress to respiratory failure and bulbar paralysis in hours or days. Sudden death, presumably from cardiac arrhythmia may occur. Bladder paresis may cause dysuria and hesitancy (1).
The central nervous system is also affected. For example, insomnia is often an early symptom. Agitation, confusion, combativeness and other acute neuropsychiatric features are common and sometimes progress to coma. Convulsions may result from porphyria itself or from hyponatremia.
Physical findings:
Tachycardia and systemic hypertension are very common. Fever is absent or mild. Signs of peritoneal irritation, such as tenderness are usually not prominent, but there is often ileus with distension and decreased bowel sounds. However, at times bowel sounds are normal or increased.
Motor weakness, which is usually symmetrical and begins proximally in the upper extremities, can be difficult to detect because common tests of strength, such as hand-grip, are not initially affected.
Clinical laboratory tests:
Leukocytosis is usually absent or mild. Hyponatremia may reflect inappropriate antidiuretic hormone secretion. Gastrointestinal or renal sodium loss may contribute.
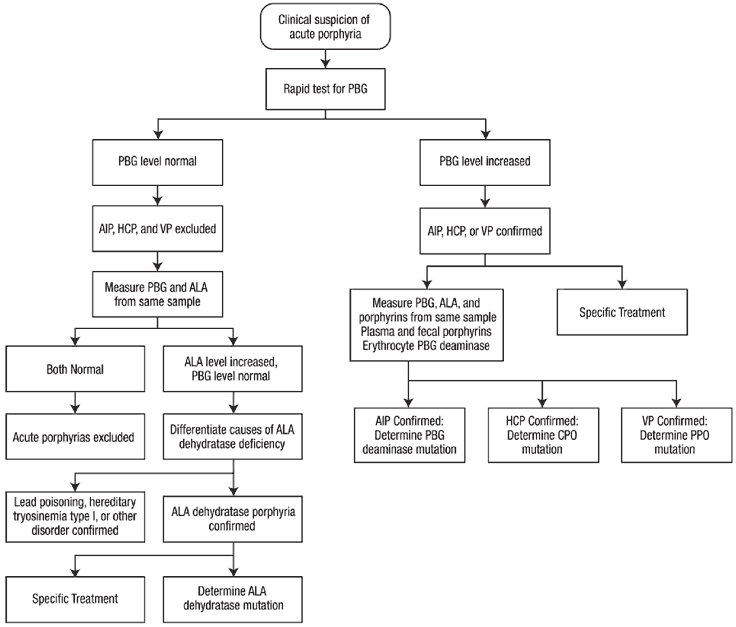
Sensitive and specific laboratory tests are available for diagnosis of acute porphyrias, especially during acute attacks, as described below.
Initial diagnosis:
The two most important diagnostic recommendations are to 1) maintain a high index of suspicion, and 2) be aware that laboratory testing is available that can readily make a diagnosis of acute porphyria or rule it out.
"Think porphyria" in patients with abdominal pain that is unexplained after an initial workup has excluded common causes such as appendicitis, cholecystitis, pancreatitis, etc. Associated findings such as reddish urine, tachycardia, hypertension, hyponatremia and proximal muscle weakness can be further indications for testing for porphyria. Other clues include use of potentially porphyrinogenic drugs such as sulfonamides, barbiturates, rifampin or metoclopramide, and premenstrual symptoms in women.
When acute porphyria is suspected, confirmation should be initiated immediately by biochemical testing. Measuring urinary PBG is most important for diagnosis of acute porphyrias. Porphobilinogen (PBG) excretion is normally 0-4 mg/day, and is approximately in the same range when expressed as mg/g creatinine or even as mg/L. In an acute attack, spot urine porphobilinogen (PBG) levels are substantially increased (20-200 mg/L).
The test should be established at the local hospital pathology clinical chemistry department. It is recommended that all medical centers provide for rapid testing for increased porphobilinogen(PBG), since sending samples out to a referral laboratory can greatly delay diagnosis and treatment. Thereby, when acute porphyria is suspected the diagnosis can be ruled in or out in a timely fashion.
If PBG is found to be elevated, treatment can be initiated without further delay. However, samples should be collected before treatment to establish the type of acute porphyria. This second-line testing should be comprehensive (Table below) - to include urinary ALA and porphyrins, plasma and fecal porphyrins, and erythrocyte porphobilinogen (PBG) deaminase, and does not require collection of 24 hour samples. DNA studies to identify the inherited mutation are important after the type of acute porphyria is established biochemically.
Recurrent attacks in a patient with proven acute porphyria are usually similar and can be diagnosed on clinical grounds, and without biochemical reconfirmation. Treatment may be initiated immediately after excluding other causes of the acute symptoms. However, the laboratory reports that were the basis for the prior diagnosis porphyria should be available for review to assure that the diagnosis was accurate.
The American Porphyria Foundation recommends known patients with hepatic porphyria wear medical alert bracelets or carry medical alert cards.
Table: Second-line testing for acute porphyrias that distinguish AIP, HCP and VP. In ADP (not shown) urine ALA (not porphobilinogen) and coproporphyrin and erythrocyte protoporphyrin are increased.
Panhematin® Therapy
The most effective therapy for the acute attack is Panhematin®. This treatment is specific, because it corrects the deficiency of regulatory heme in the liver and down-regulates ALAS. Glucose loading has a similar effect, but is much less potent and effective and should be used only for mild attacks.
The standard Panhematin® treatment course is 3-4 mg/kg by vein once daily for 4 days. If the diagnosis is confirmed, the first dose can be given in the ER. Panhematin® (Recordati Rare Diseases) is the only hemin therapy available in the United States. It is a lyophilized preparation of hydroxyheme (hematin) and is FDA approved for treating acute porphyrias. To order Panhematin®, contact Recordati Rare Diseases at: www.recordatirarediseases.com or 866-654-0539
Recordati Rare Diseases will express ship Panhematin® to the pharmacy on request. Heme arginate is a hemin preparation available in Europe and South Africa and is administered at the same dose.
Panhematin® can cause phlebitis at the site of infusion, which may compromise venous access. Experts recommend that lyophilized Panhematin® be reconstituted with human albumin rather than sterile water, which enhances its stability and decreases the chance of phlebitis. The method for this has been published (5). In brief, the authors recommended reconstituting a 313 mg vial of Panhematin® with 132 ml of 25% human serum albumin, then swirling 15-20 times to mix. The amount required is then withdrawn into a syringe using a 5-micron filter needle, and injected into a 150 ml sterile bottle prior to IV infusion. This must be placed in an amber bag to protect from light, and used within one hour of preparation, in part because the solution contains no bacteriostatic agents. The concentration of hemin as heme-albumin is 2.4 mg/mL, which is used to calculate the volume needed to deliver a dose of 3-4 mg/kg. The dose is generally infused by piggyback to an intravenous line that is infusing 0.9% sodium chloride at a moderate rate. The dose should be infused over a period of at least 60 minutes or at a rate that should not exceed 1 mL/min (5). Panhematin® treatment can be continued if recovery is not evident within 4 days.
Complications of IV Panhematin® therapy other than local phlebitis include fever, aching, malaise, and hemolysis. Rarely, there have been reports of transitory circulatory collapse and, after excessive dosing, renal failure. Panhematin® has been given safely in pregnancy.
Patients should be monitored closely and treated for pain and other symptoms, and for complications such a respiratory impairment, hyponatremia and neuropsychiatric manifestions. Monitoring in an intensive care unit is generally advisable (1). Levels of ALA and porphobilinogen (PBG) can be monitored, and should decrease rapidly with Panhematin® treatment. However, this biochemical response does not necessarily predict a clinical response, which will depend on how advanced the neuropathic manifestations were before treatment was started.
Supportive and Symptomatic Treatment:
Harmful drugs should be stopped immediately and avoided during treatment. For example, metoclopramide should not be given. Information on harmful medications is found at the APF (porphyriafoundation.org/drug_database/) and EPI websites (www.drugs-porphyria.org) as searchable databases that are updated periodically (1).
Intravenous glucose loading (at least 3 L of 10% glucose daily) should be used only for mild attacks (pain controlled with little or no narcotic analgesics, and without hyponatremia, motor weakness, etc.) or while waiting for Panhematin® to become available. During treatment with hemin, large volumes of 10% glucose are not needed, which helps prevent dilutional hyponatremia. Nutrition support can be given using a central line and more concentrated solutions if needed during a prolonged attack.
Hyponatremia, hypomagnesemia and other electrolyte imbalances should be corrected and monitored. Safe drugs include narcotic analgesics for pain and phenothiazines for nausea, vomiting, anxiety or agitation. Beta-adrenergic blocking agents can be used to control the tachycardia and systemic arterial hypertension in patients without hypovolemia. Seizure precautions are recommended, especially in patients who develop hyponatremia. Treatment of seizures is difficult, since most commonly used anticonvulsants are harmful in acute porphyria. Gabapentin, benzodiazepines and vigabatrin are considered safe. Patients who experience seizures during an acute attack seldom require prolonged anticonvulsant therapy.
Patient education, genetic counseling, a medic alert bracelet or card, and consultation with a porphyria expert for additional advice and follow-up is recommended after recovery from an acute attack. The risk of hepatocellular carcinoma is increased in the acute porphyrias (6,7). Therefore, it is currently recommended that patients undergo screening by liver imaging for early detection at least yearly after age 50, especially if porphobilinogen (PBG) remains elevated.
The American Porphyria Foundation is a valuable source of additional advice. Patients should be encouraged to join and support this association.
Attacks recur in some patients, even if they avoid drugs and other factors that are known to cause exacerbations. Patients with frequent premenstrual attacks may benefit from treatment with a gonadotropin-releasing hormone analogue (8). A single infusion of Panhematin® once or twice weekly may prevent noncyclic attacks in some patients, but experience is limited. Some patients treated repeatedly with hemin develop iron overload, which can be assessed by serum ferritin measurement, and treated by phlebotomies. Liver transplantation has been effective in a few patients with recurrent attacks that were not responsive to other interventions (2). Gene therapy may become available in the future.
Some patients develop chronic symptoms, including chronic pain syndrome and severe depression with an increase risk of suicide. These patients require careful supportive management, and their symptoms may improve over time.
*Please note that symptoms and treatment of HCP and VP are similar.
1) Anderson KE, Bloomer, JR Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR and Desnick RJ. Recommendations for the Diagnosis and Treatment of the Acute Porphyrias. Ann Intern Med 2005; 142:439-450
2) Seth AK, Badminton MN, Mirza D, Russell S, Elias E, Liver transplantation for porphyria: Who, when, and how? Liver Transplantation. 2007;1219-1227
3) Deacon AC, Peters TJ, Identification of acute porphyria: evaluation of a commercial screening test for urinary porphobilinogen. Ann Clin Biochem. 1998;35:726-32
4) Blake D, McManus J, Cronin V, Ratnaike S. Fecal coproporphyrin isomers in Hereditary Coproporphyria. Clin Chem. 1992;38:96-100
5) Anderson KE, Bonkovsky HL, Bloomer JR and Shedlofsky SI. Reconstitution of he- matin for IV injection. Ann Intern Med 2006; 144:537-38
6) Andersson C, Bjersing L, Lithner F. The epidemiology of hepatocellular carcinoma in patients with Acute Intermittent Porphyria. J Intern Med. 1996; 240:195-201
7) Andant C, Puy H, Faivre J, Deybach JC. Acute hepatic porphyrias and primary liver cancer [Letter]. N Eng J Med 1998; 338:1853-4
8) Anderson KE, Spitz IM, Bardin CW, Kappas A. A gonadotropin releasing hormone analogue prevents cyclical attacks of porphyria. Arch Intern Med. 1990; 150: 1469-74



