FOR SUPPORT, CALL 1-866-APF-3635 (273-3635)
AURORA study designed to evaluate bitopertin as a potential disease-modifying treatment for adults with EPP in the United States.
Aurora study designed to assess changes in protoporphyrin IX levels, safety, tolerability, photosensitivity and other measures in a double-blind, placebo-controlled setting; SEE https://www.discmedicine.com/news/disc-medicine-initiates-aurora-a-phase-2-clinical-study-of-bitopertin-in-adults-with-erythropoietic-protoporphyria-epp/
Also see: https://clinicaltrials.gov/ct2/show/NCT05308472
Disc Presents Positive Initial Data from Phase 2 BEACON Trial of Bitopertin for EPP: https://ir.discmedicine.com/news-releases/news-release-details/disc-presents-positive-initial-data-phase-2-beacon-trial
Disc medicine has launched an Health Care Professional (HCP) focused site offering dedicated resources to support clinical practice, including:
Disc Medicine is committed to deepening the understanding of these conditions, please visit the new sites and explore what’s available:
lthcare Professionals: for Erythropoietic Protoporphyria & X-Linked Protoporphyria http://www.ppixiswhyhcp.com
Another exciting educational video series for EPP management can be viewed at www.medlive.com/v/EPP EPP experts , Dr Bruce Wang from the University of California , San Francisco and Dr. Gayle Ross from the Royal Melbourne Hospital in Melbourne , Australia provide presentations on the Erythropoietic Prototoporphyria (EPP): Phototoxic light triggered reactions and systemic complications associated with the accumulation of PPIX.
Disc Medicine is proud to announce our new campaign to raise awareness for Erythropoietic Protoporphyria including X-Linked Protoporphyria. The campaign was built specifically for people living with EPP and XLP and their caregivers and is intended for US residents. It is here to help with
In addition to the Patient and Caregiver site, Disc also launched an Health Care Professional (HCP) focused site offering dedicated resources to support clinical practice, including:
Disc Medicine is committed to deepening the understanding of these conditions, please visit the new sites and explore what’s available:

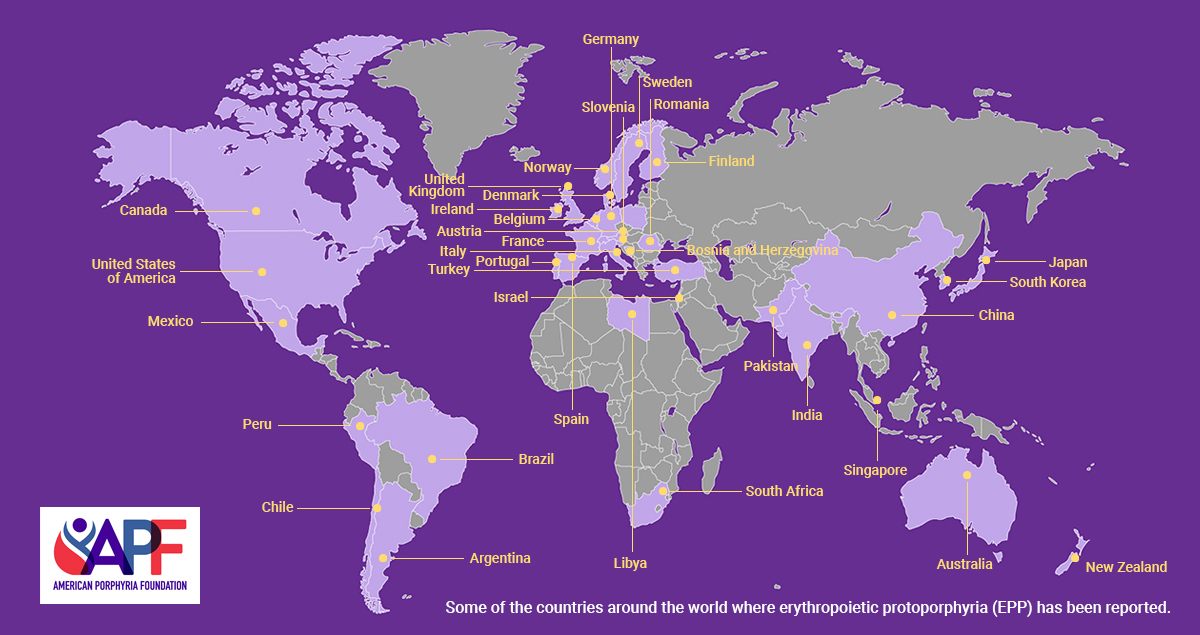
Erythropoietic Protoporphyria (EPP) is a rare inherited metabolic disorder characterized by a deficiency of the enzyme ferrochelatase (FECH). Due to abnormally low levels of this enzyme, excessive amounts of protoporphyrin accumulate in the bone marrow, blood plasma, and red blood cells. Protoporphyrin is excreted by the liver into the bile, after which it enters the intestine and is excreted in the feces. It is not soluble in water so is not excreted in the urine. The major symptom of this disorder is hypersensitivity of the skin to sunlight and some types of artificial light, such as fluorescent lights (photosensitivity). After exposure to light, the skin may become itchy and red. Affected individuals may also experience a burning sensation on their skin. The hands, arms, and face are the most commonly affected areas. Some people with Erythropoietic Protoporphyria may also have complications related to liver and gallbladder function.
Erythropoietic Protoporphyria is characterized by abnormally elevated levels of protoporphyrin IX in erythrocytes (red blood cells) and plasma (the fluid portion of circulating blood), and by sensitivity to visible light that is usually noticed in early childhood and occurs throughout life. EPP can result either from mutations of the ferrochelatase gene (FECH), or less commonly the delta-aminolevulinic acid synthase-2 gene (ALAS2). When EPP is due to an ALAS2 mutation it is termed X-linked protoporphyria (XLP), because that gene is found on the X chromosome.
EPP and XLP, combined, are the third most common Porphyria, with an incidence of possibly 2 to 5 per 1,000,000. They are the most common Porphyria in children. EPP is caused by a lack of the enzyme, ferrochelatase due to mutations in the FECH gene.
Erythropoietic Protoporphyria affects males and females in equal numbers. It is estimated that the disorder occurs in about 1 in about 74,300 individuals. The onset of symptoms affecting the skin usually occurs in infancy; however, in some cases, onset may not occur until adolescence or adulthood.
X-linked Protoporphyria affects males and females. However, males usually develop a severe form of the disorder while females with an ALAS2 mutation may range from having no symptoms (asymptomatic) to developing a severe form of the disorder. The exact incidence or prevalence of X-linked Protoporphyria is unknown. The disorder has only been reported in the medical literature in a handful of families in Europe, South Africa and Japan.
There is an increased risk of gallstones, which contain protoporphyrin. Excess protoporphyrin can also cause liver damage. Less than 5% of EPP patients’ severe liver damage and a condition caused protoporphyric hepatopathy that sometimes requires liver transplantation.
Erythropoietic Protoporphyria is a rare disorder inherited as an autosomal dominant genetic trait with poor penetrance. Human traits, including the classic genetic diseases, are the product of the interaction of two genes, one received from the father and one from the mother.
When EPP is due to a FECH mutation the inheritance is described as autosomal recessive. It is most common to find that one severe mutation is inherited from one parent and another weak mutation inherited from the other parent. The weak mutation is quite common in normal Caucasians, rare in Blacks and even more common in Japanese and Chinese populations. This mutation is sometime referred to as “hypomorphic” because it results in formation of a less than normal amount of ferrochelatase. But is does not cause EPP unless it is paired with a severe mutation. The severe mutation is characteristic for an EPP family and is present in all affected individuals. “Carriers” of the severe mutation are not affected because they do not have the weak mutation. Affected individuals and unaffected carriers can transmit the severe mutation to the next generation. Some of their children will have EPP if the other parent has a copy of the weak mutation. Rarely, the weak mutation is absent in an EPP family and two severe mutations are found, with at least one producing some ferrochelatase.
In XLP, mutations of the ALAS2 gene, which is found on the X chromosome, causes an increase in the production of the enzyme ALAS2 in the bone marrow. Several of these “gain of function” mutations have been described in different XLP families. In XLP protoporphyrin production exceeds that needed for heme and hemoglobin formation. Like hemophilia and other X linked genetic diseases, XLP is more common in men. Women have two X chromosomes and are usually not affected because they have a normal as well as a mutated ALAS2 gene. Men have only one X chromosome and will be affected if they inherit an ALAS2 mutation. Women with an ALAS2 mutation will, on average, pass that mutation to half of their daughters (who will usually be unaffected carriers) and to half of their sons (who will be affected).
In dominant disorders, a single copy of the disease gene (received from either the mother or father) will be expressed “dominating” the other normal gene and resulting in the appearance of the disease. The risk of transmitting the disorder from affected parent to offspring is 50 percent for each pregnancy regardless of the sex of the resulting child. The risk is the same for each pregnancy.
The symptoms of Erythropoietic Protoporphyria develop due to excessive levels of a chemical called protoporphyrin that accumulates in certain tissues of the body (i.e., the plasma, red blood cells, and the liver). Excessive protoporphyrin levels occur as the result of abnormally low levels of the enzyme ferrochelatase (FECH).
Swelling, burning, itching, and redness of the skin may appear during or after exposure to sunlight, including sunlight that passes through window glass. This can cause mild to severe burning pain on sun-exposed areas of the skin. Usually, these symptoms subside in 12 to 24 hours and heal without significant scarring. Blistering and scarring are characteristic of other types of cutaneous Porphyria but are unusual in EPP. Skin manifestations generally begin early childhood and are more severe in the summer.
The most common symptom of Erythropoietic Protoporphyria is hypersensitivity of the skin to sunlight and some types of artificial light (photosensitivity), with pain, itching, and/or burning of the skin occurring after exposure to sunlight and occasionally to fluorescent light. Affected individuals may also exhibit abnormal accumulations of body fluid under affected areas (edema) and/or persistent redness or inflammation of the skin (erythema). In rare cases, affected areas of the skin may develop sac-like lesions (vesicles or bullae), scar, and/or become discolored (hyperpigmentation) if exposure to sunlight is prolonged. However, scarring and/or discoloring of the skin is uncommon and rarely severe. These affected areas of skin may become abnormally thick. In addition, in some cases, affected individuals may also exhibit malformations of the nails. The severity and degree of photosensitivity is different from case to case. Photosensitivity is often seen during infancy; however, in some cases, it may not occur until adolescence or adulthood.
In some affected individuals, the flow of bile through the gallbladder and bile ducts (biliary system) may be interrupted (cholestasis) causing gallstones (cholelithiasis) to form. In turn, such stones can cause obstruction and/or inflammation of the gallbladder (cholecystitis). Rarely, affected individuals may also develop liver damage that, in very severe cases, may lead to liver failure requiring transplantation.
Symptoms usually start in childhood, but diagnosis is often delayed since blistering is not common and, because the porphyrins are insoluble, they usually escape detection on urinanalysis. The diagnosis is made upon finding increased levels of the protoporphyrin in the plasma or red blood cells.
EPP should be suspected in anyone with non-blistering photosensitivity especially when it is prolonged and beginning in childhood. It is easy to make a diagnosis, or rule it out, once it is suspected.
The diagnosis of EPP is established by finding an abnormally high level of total erythrocyte protoporphyrin and showing that this increase is mostly free protoporphyrin rather than zinc protoporphyrin. There is considerable confusion about which test to order. Sometimes laboratories have measured only zinc protoporphyrin and reported results incorrectly as “protoporphyrin” or “free erythrocyte protoporphyrin (FEP)”. Laboratories that measure total erythrocyte protoporphyrin, free protoporphyrin and zinc protoporphyrin and report results reliably are:
Porphyrins are almost always elevated in plasma in EPP but may be normal in mild cases. Fecal porphyrins may be normal or increased.
A diagnosis of X-linked protoporphyria may be made through blood tests that can detect markedly increased levels of metal-free and zinc-bound protoporphyrins within red blood cells (erythrocytes). A higher ratio of zinc-bound protoporphyrin to metal-free protoporphyrin can differentiate X-linked protoporphyria from EPP.
Molecular genetic testing can confirm a diagnosis of X-linked protoporphyria by detecting mutations in the ALAS2 gene (the only gene known to cause this disorder).
An experienced biochemical laboratory can usually distinguish between patients with EPP and XLP, because the former has much less zinc protoporphyrin in their erythrocytes. This can be explained because in the marrow the enzyme ferrochelatase not only normally makes heme (iron protoporphyrin) from protoporphyrin and iron, but can also make zinc protoporphyrin, especially when excess protoporphyrin is present or iron is deficient. However, this does not replace DNA studies.
Rarely, EPP develops in adults in the presence of a bone marrow disorder such as polycythemia vera, and is due to expansion of a clone of red blood cell precursors in the marrow that is deficient in ferrochelatase.
DNA studies are important for confirming the diagnosis of EPP and XLP and for genetic counseling. This should be completed first in a person known to have the disease, and the information about the mutations in that individual used to guide testing of family members.
The diagnosis of Erythropoietic Protoporphyria (EPP) may be made by a thorough clinical evaluation, characteristic physical findings, and specialized laboratory tests. EPP is usually diagnosed during infancy or early childhood, due to characteristic skin symptoms. The diagnosis may be confirmed by testing the red blood cells (erythrocytes) for increased levels of protoporphyrin.
SCENESSE® is a prescription medication that contains the active substance afamelanotide. Afamelanotide is used to increase tolerance to the sun and light in adults with a confirmed diagnosis of erythropoietic protoporphyria (EPP).
SCENESSE® (pronounced “sen-esse”) acts by increasing the levels of eumelanin in the skin, shielding against UV radiation (UVR) and visible light, including sunlight. Afamelanotide is a synthetic form of a hormone called alpha-melanocyte stimulating hormone (?-MSH). Afamelanotide works in a way similar to the natural hormone, by making skin cells produce eumelanin which is a brown-black type of melanin pigment in the skin. By increasing the amount of eumelanin and acting as an antioxidant, SCENESSE® can help to reduce the sensitivity of the skin to sunlight and artificial UV light sources.
Implant is given subcutaneously by a trained health care professional.
Click here to continue reading about SCENESSE®
Protection from sunlight is the mainstay of management of EPP, and this is necessary throughout life. Disease severity and porphyrin levels in erythrocytes and plasma probably remain high and relatively constant throughout life in EPP. However, this has been little studied and more longitudinal observations are needed. Life style, employment, travel and recreation require adjustment in order to avoid painful reactions to sunlight and even from exposure to fluorescent lighting. For these reasons EPP can substantially affect quality of life.
Protective clothing, including broad-brimmed hats, long sleeves, gloves and trousers (rather than shorts), is beneficial. Several manufacturers specialize on clothing made of closely woven fabrics for people with photosensitivity.
In an occasional patient, protoporphyrin causes liver problems, so monitoring liver function is important. EPP patients should also not use any drug or anesthetic which causes cholestasis (slowing down bile flow) and should also avoid alcohol. Women should avoid medications containing estrogen (birth-control pills, hormone replacement therapy), and men should avoid testosterone supplements, as these substances can also have deleterious effects on the liver of a person with EPP.
Yearly monitoring. Testing to include erythrocyte total protoporphyrin, plasma porphyrin, complete blood counts, ferritin and liver function tests should be done yearly. Porphyrin levels are expected to be stable and liver tests to remain normal. EPP patients may have evidence of iron deficiency, and an iron supplement may be advisable if the serum ferritin is below about 20 ng/mL.
Vitamin D. Because they avoid sunlight, EPP patients are likely to be deficient in vitamin D. A vitamin D supplement with calcium is recommended for bone health.
Liver protection. It is important to avoid other causes of liver disease that might promote the development of liver complications from EPP. Patients should avoid alcohol and other substances that might damage the liver, including many herbal preparations, and be vaccinated for hepatitis A and B.
Surgical lights. Strong operating room lights can cause photosensitivity of the skin and even surfaces of internal organs. Flexible membrane filters, such as CL5-200-X from Madico Co., are available to cover surgical lights and offer some protection. This is especially important in EPP patients with liver failure, which causes even greater increases in protoporphyrin levels and photosensitivity.
Drugs. Drugs that are harmful in other Porphyrias are not known to make EPP worse but are best avoided as a precaution. This may include estrogens and other drugs that might reduce bile formation. A short course of a non-steroidal anti-inflammatory drug can provide some pain relief after an episode of photosensitivity but can cause ulcerations of the digestive track especially with prolonged use.
Laser treatment. Laser treatments for hair removal or eye surgery have not been a problem in EPP people. But the doctor should be made aware of the diagnosis, and that laser output between 400 and 650 nanometers might be harmful. Before hair removal treatment, the doctor may irradiate a small area of the skin to be treated for the length of time it will take to do the hair removal to ascertain if the patient would react within the period of time that a reaction to sunlight would be expected in that patient.
Children with EPP. Please see the Shadow Jumpers area of this website.
Beta-Carotene. In Erythropoietic Protoporphyria, a high potency form of oral beta-carotene (Lumitene, Tishcon) may be given to improve an affected individual's tolerance of sunlight. In some cases, the drug cholestyramine may be given to alleviate skin symptoms and lower the protoporphyrin levels in the body.
Iron. When iron deficiency is present, iron supplements may be given.
Genetic counseling will be of benefit for affected individuals and their families. Other treatment is symptomatic and supportive.
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
Active Clinical Trial:
Mitsubishi Tanabe MT-7117 Clinical Trial
Phase 3 Clinical Trial
Participants with EPP or XLP are needed
Trial Description: A Phase 3, Global, Multicenter, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate Efficacy, Safety, And Tolerability of MT-7117 in Subjects with Erythropoietic Protoporphyria (EPP) or X-Linked Protoporphyria (XLP)
Treatment: Oral medication (pills), once daily in the morning with or without food Duration: 26 weeks plus optional 26 week double-blinded extension.
Endpoint: Increased pain free light exposure in adults and adolescents with a history of phototoxic reactions from EPP or XLP.
This clinical trial is now closed for enrollment. Please contact the American Porphyria Foundation for additional information.
Phone: 866-APF-3635 or Email: general@porphyriafoundation.org
See Member Stories on Erythropoietic Protoporphyria (EPP) and X-Linked Protoporphyria (XLP)



