FOR SUPPORT, CALL 1-866-APF-3635 (273-3635)
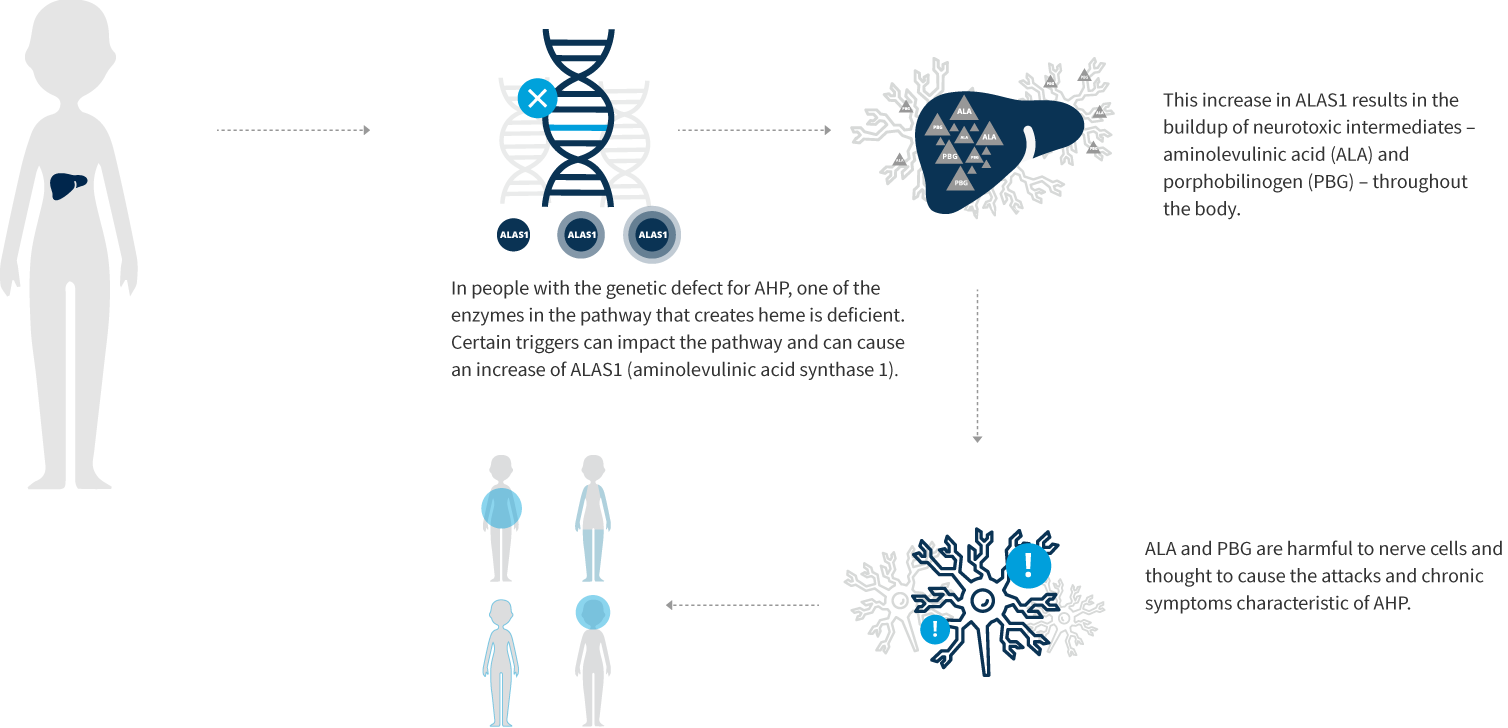
Porphyria is a family of rare, genetic diseases characterized by potentially life-threatening attacks and, for some patients, chronic debilitating symptoms that negatively impact daily functioning and quality of life. Porphyria includes eight subtypes, and each has unique manifestations, making an accurate diagnosis even more challenging. Four of these subtypes, a family of diseases referred to as acute hepatic porphyria (AHP), result from a genetic defect leading to deficiency in one of the enzymes of the heme biosynthesis pathway in the liver: acute intermittent porphyria (AIP), variegate porphyria (VP), hereditary coproporphyria (HCP), and ALAD-deficiency porphyria (ADP).
In the United States and Europe, approximately 5,000 people experience one or more attacks annually, and approximately 1,000 people suffer frequent and severe attacks. Common treatment approaches for AHP include carbohydrate loading, intravenous (IV) glucose, and IV hemin, which can be administered during an attack or sometimes between attacks (prophylactically). However, hemin is not currently approved for the prophylactic treatment of AHP. Currently, there are no treatments approved to prevent debilitating attacks and treat the chronic symptoms of AHP.
Affected individuals often experience some combination of the following symptoms: