Other Important Treatment Considerations for the Acute Porphyrias
Saturday - December 24, 2016 @ 06:30:13
Other Important Treatment Considerations for the Acute Porphyrias
Hospitalization is often necessary for acute attacks. Medications for pain, nausea, and vomiting, as well as close observation are generally required.
A high intake of glucose or other carbohydrates can help to suppress disease activity and can be given by vein or by mouth.
Attacks are often precipitated by low intake of carbohydrates and calories in an attempt to lose weight. Thus dietary counseling is very important.
Premenstrual attacks often resolve quickly with the onset of menses. Hormone manipulations may prevent such attacks.
Acute porphyria is particularly dangerous if the diagnosis has not been made and if harmful drugs are administered. The prognosis is usually good if the disease is recognized and if treatment and preventive measures are begun before severe nerve damage has occurred. Although symptoms usually resolve after an attack, some patients develop chronic pain. Nerve damage and associated muscle weakness can improve over a period of months or longer after a severe attack. Mental symptoms may occur during attacks but are usually not chronic.
Wearing a MedicAlert bracelet is advisable for patients who have been diagnosed with porphyria.
Bibliography:
Anderson KE. The porphyrias. In: Goldman L, Ausiello D, eds. Cecil Textbook of Medicine. Philadelphia, Pa: WB Saunders Co; 2004: chap. 223.
Bonkovsky HL, Healey BS, Lourie AN, Gerron GG. Intravenous heme-albumin in acute intermittent porphyria: evidence for repletion of hepatic hemoproteins and regulatory heme pools. Am J Gastroenterol. 1991;86:1050-1056.
Data on file, Recordati Rare Diseases, Inc.
Reimbursement
What is the Recordati Rare Diseases Reimbursement Support Program? The Recordati Rare Diseases Reimbursement support Program is a free service available to patients, caregivers, medical billing staff, healthcare providers, and others who have questions about insurance coverage and reimbursement-related issues.
What does the Recordati Rare Diseases Reimbursement Support Program offer? The Program offers comprehensive assistance for insurance-related issues:
Billing issues Contact the hotline for assistance obtaining appropriate billing codes required on medical claims or if you need additional documentation to submit with your claims
Insurance verification The hotline representative will contact your insurance company to determine in advance how it will pay for the Recordati Rare Diseases product or related procedure in which you are interested
Prior authorization support The hotline representative can help facilitate the prior authorization process by determining requirements, coordinating paperwork, and following up on the final decision
Insurer education Representatives who staff the hotline can help educate insurers about Recordati Rare Diseases products to expedite coverage and payment
Policy monitoring The Program monitors public and private payor coverage policies to ensure you have the most up-to-date coverage information. Representatives will also answer any questions you have about insurance coverage and reimbursement related to Recordati Rare Diseases products.
If you are unable to contact us during our regular hours (M - F between 9 am and 5 pm eastern time), simply leave a message with your name, a phone number where you can be reached, and your question. A reimbursement professional will return your phone call within one business day.
For other Panhematin related questions:
Dial 911 if you are experiencing a medical emergency.
Global Genes is hosting the first-ever RARE in the Square event in San Francisco! See the announcement below for details! A Unique Partnering Opportunity for Rare Disease Innovators January 9th 11th, 2017
Wednesday - December 21, 2016 @ 09:30:14
Global Genes is hosting the first-ever RARE in the Square event in San Francisco! See the announcement below for details!
A Unique Partnering Opportunity for Rare Disease Innovators
January 9th 11th, 2017
A Catalyzing RARE Community Event Global Genes is pleased to announce the first-ever RARE in the Square (RITS) hosted January 9 -11, 2017 in San Franciscos Union Square. The inaugural RITS will coincide with the JP Morgan Healthcare Conference (JPM), creating the opportunity for rare disease focused companies and organizations to network at the epicenter of JPM-related activities in a unique, one-of-a-kind space.
The focus on rare disease innovation continues to increase at the JP Morgan Healthcare Conference. The rare community involvement and representation at the JPM Healthcare Conference is growing annually, comments Nicole Boice, CEO and Founder of Global Genes. However, the rare disease communitys true presence and impact hasnt been represented at its fullest until this year. That is why we are so thrilled for the opportunity to partner in conjunction with JPM to provide the much-needed collaborations and visibility in support of rare disease innovation.
Located in the heart of San Francisco and across the street from The Westin and JPM conference, Union Square provides a unique, distinctive and convenient location. This space lends RITS a platform where investors, industry partners and patient community leaders can connect, network or even just relax amongst fellow rare enthusiasts.
Patients and their families are the most powerful drivers of the orphan drug development process. They allow us to understand the purpose, the urgency. They allow us to focus and to explain to the many parties involved. They allow us to acquire the passion to succeed and to never give upâ?¦ Nothing is more important. Henri Termeer, Former Chairman, President and CEO, Genzyme Corporation
Alnylum Pharmaceutical UPDATE
Monday - December 19, 2016 @ 09:30:07
At the request of Desiree Lyon:
Alnylum Pharmaceutical has nearly completed Phase I of the new porphyria drug ALN-AS1 with very promising results. Alnylum specializes in RNA interference (RNAi) treatments which target specific cells that are the root cause of various diseases. Alnylum targets rare diseases that currently have limited treatments, especially blood disorders such as hemophilia and acute porphyria.
Researchers and doctors worldwide agree that porphyria is an extremely rare genetic disease in which the patient does not properly produce heme, an important molecule in blood, which causes a build up of naturally occurring neurotoxins ALA and PBG. These neurotoxins build up in the liver and eventually spill into other areas of the body causing a variety of symptoms including severe abdominal pain, nausea, vomiting, constipation, diarrhea, painful body aches, muscle weakness, paralysis and in some cases psychosis, encephalopathy and death.
ALN-ASI or Givosiran (gi-VOH-sir-an) targets liver cells that are responsible for heme production and the root cause of acute porphyria. By attaching an RNAi to a sugar molecule, Givosiran goes directly to the liver cells where it slows down heme production. Early results show a 74% decrease in ALA and PBG buildup and a 74% decrease of porphyria attacks in patients. There has only been one adverse event reported and that is skin irritation at the injection site. This is a very common occurrence with any injection treatment. Givosiran is injected once every 4 weeks using the same procedure diabetics use to inject insulin. Potentially, Givosiran will be able to be self-administered at home and not require a doctor's appointment.
The results have been so promising, Alnylum believes it can safely bypass Phase II testing and will be seeking this exception from the Food and Drug Administration (FDA) in early 2017 in hopes of starting large scale human trials (Phase III) by the end of 2017.
Alnylum did extensive patient research before developing the drug and discovered how limited current treatments are for porphyria patients. Panhematin has been the most widely used treatment for acute porphyria patients and has been in use for over thirty years. It requires 3 to 11 intravenous treatments over consecutive days during acute attacks or can be used prophylactic requiring patients to receive the treatment anywhere from once a month to twice a week. Treatments take up to an hour and usually require patients to have an intravenous access port surgically installed. It is extremely exciting to see a company take a modern medicine approach to treat a disease that affects nearly 5,000 patients worldwide.
Alnylum believes Givosiran has the potential to greatly reduce or eliminate the rarely spoken about chronic aspects of porphyria, the frequency and severity of attacks, as well as reducing the risk of liver cancer which is common in long time sufferers of porphyria.
The American Porphyria Foundation (APF) has played an important role in helping Alnylum develop Givosiran. By creating the Porphyria Consortium, Alnylum was able to move quickly through the research and expert recruitment phase of the drug development. The APF has assisted Alnylum recruit patients as well as asymptomatic high excreters of ALA and PBG who volunteered for Phase I. In addition, The APF has sent porphyria patients to meet with the development teams at Alnylum to provide insight into battling porphyria on a daily basis as well as providing advice to help recruit patients for Phase III and address concerns they may have. Finally, the APF will play a critical role in assisting the recruitment of the more than 100 patients worldwide to participate in Phase III.
Are you prepared for a widespread disease or epidemic?
Friday - December 16, 2016 @ 10:30:14
How to Reduce Your Risk During an Epidemic
In 2014, the Ebola virus spread rapidly throughout West Africa, making headlines around the world.
How did you help people to understand the danger they faced?
We sought to dispel fear and confusion by making special presentations at public places. In those presentations, we explained how the virus spreads and warned against unsafe practices.
What practical steps were taken?
We used infrared thermometers to check the temperature of those arriving for meetings at our places of worship. Everyone carefully avoided unnecessary physical contact, such as shaking hands or hugging, and washed their hands frequently throughout the day. At strategic locations throughout the communities, handwashing stations were set up with a bleach solution.
What happened if someone showed symptoms?
Authorities were notified. Any individuals who had been in contact with an Ebola victim, had attended a funeral of a victim, or showed symptoms kept themselves isolated for 21 days, the commonly accepted maximum incubation period for the Ebola virus.
Protect Yourself From Disease
Wednesday - December 14, 2016 @ 09:30:00
Protect Yourself From Disease
MANY ancient cities were protected by massive walls. If an enemy breached just a small section of a wall, the safety of the entire city was at risk. Your body is like a walled city. How you care for your defenses has much to do with how healthy you are. Consider five elements that can expose you to disease and how you can put up the best possible defenses.
1 WATER
THE THREAT: Harmful organisms can march straight into your body by way of contaminated water.
YOUR DEFENSE: The best defense is to protect your water supply from contamination. If you know that your water supply is contaminated or suspect that it is, you can treat the water at home to make it safe.* Store potable water in a closed vessel, and dispense it hygienically with a clean ladle or through a tap. Never put your hands into a clean water supply. If possible, you should try to live in a community that properly disposes of human waste so that it does not contaminate local water sources.
2 FOOD
THE THREAT: Harmful organisms can be present in or on your food.
YOUR DEFENSE: Contaminated food may look fresh and nutritious. So get into the habit of thoroughly washing all fruits and vegetables. Ensure that food utensils, kitchen surfaces, and your hands are clean when preparing or serving food. Some foods require cooking at a certain temperature in order to destroy dangerous microbes. Beware of food that is discolored or has an unpleasant odor or tastesigns that an army of microorganisms could be waiting for you. Refrigerate unused food as soon as possible. Avoid preparing food for others when you are sick.*
3 INSECTS
THE THREAT: Some insects can infect you with the harmful microorganisms that live inside them.
YOUR DEFENSE: Limit contact with disease-carrying insects by staying indoors when they are active or by wearing protective clothing, such as long sleeves and long trousers. Sleep under treated insect nets, and use personal insect repellent. Eliminate containers of stagnant water where mosquitoes could breed.*
4 ANIMALS
THE THREAT: Microbes that live harmlessly inside an animal can threaten your health. If you are bitten or scratched by a pet or another animal or exposed to its feces, you could be at risk.
YOUR DEFENSE: Some people choose to keep their animals outside the house to minimize contact with them. Wash your hands after touching a domestic animal, and avoid all contact with wild animals. If you are bitten or scratched, wash the wound thoroughly and seek a doctors advice.*
5 PEOPLE
THE THREAT: Some germs can invade your body by riding on tiny droplets in someones cough or sneeze. They can also spread through skin contact, such as hugging or shaking hands. Microorganisms from other people may lurk on such items as doorknobs, handrails, telephones, remote controls, or computer screens and keyboards.
YOUR DEFENSE: Do not share personal items, such as razors, toothbrushes, or towels. Avoid contact with body fluids from animals or from other people, including blood and products derived from blood. And do not underestimate the benefits of washing your hands thoroughly and frequently. It is perhaps the most effective way you can stop the spread of infection.
If possible, stay home when you are sick. The U.S. Centers for Disease Control and Prevention recommends that you cough or sneeze into a tissue or your sleeve, but not into your hands.
An ancient proverb states: The shrewd one sees the danger and conceals himself. How true are those words today in a world plagued with potentially dangerous diseases! So inform yourself by consulting local health services, and conceal yourself from danger by practicing good hygiene. Bolster your defenses, and reduce the risk of disease!
DiseaseHow to Reduce The Risk
Friday - December 9, 2016 @ 09:30:03
DiseaseHow to Reduce The Risk
Every day your body wages war against enemies that are silent and unseen but potentially deadly. Foreign invaders, such as bacteria, viruses, and parasites, threaten your health.* You are not likely to be aware of those battles because your immune system repels or destroys most of the invaders before the onset of symptoms. Sometimes, however, the harmful germs gain the upper hand. If so, you may need to bolster your defenses with medicine and other treatments.
For thousands of years, people knew virtually nothing about the dangers of microscopic or other small harmful organisms. However, when 19th-century scientists confirmed the link between germs and disease, we became better equipped to defend ourselves. Medical researchers have since eliminated or greatly reduced the threat of some infectious diseases, including smallpox and polio. Recently, however, others, such as yellow fever and dengue, have made a comeback. Why? Consider these factors:
Every year, millions of people travel around the globe, often transporting disease-causing agents. According to an article in the journal Clinical Infectious Diseases, virtually all of the contagious virulent infections can be spread by international travelers.
Some bacteria have developed resistance to antibiotics. The world is heading towards a post-antibiotic era, in which common infections . . . can once again kill, states the World Health Organization.
Civil unrest and poverty often hinder government efforts to control the spread of disease.
Many people lack practical knowledge of how to prevent disease.
Despite these disturbing trends, there is much you can do to protect yourself and your family. The following will show that, even if you live in a developing land, simple and effective strategies may be within your reach.
EPP Online docket Question's for FDA
Monday - December 5, 2016 @ 09:00:22
Following the FDA meeting for EPP that was held in October, the FDA opened an online public docket for EPP people to submit comments. This docket is open to attendees of the meeting AND those that were unable to attend in person. The FDA is particularly interested in hearing patients' perspectives on the questions discussed during the workshop. These questions are pasted below for your reference. If you have any questions, please email meghana.chalasani@fda.hhs.gov.
Comments MUST be submitted by December 24, 2016. Submit your comments through this website:
1. Of all the symptoms that you experience because of your condition, which 1-3 symptoms have the most significant impact on your daily life? (Examples may include itching, burning, pain, scarring, etc.)
2.Are there specific activities that are important to you but that you cannot do at all or as fully as you would like because of your condition? (Examples of activities may include daily hygiene, work and school performance, participation in sports or social activities, etc.)
3.How have your condition and its symptoms changed over time?
4.What are you currently doing to manage your condition or its symptoms? (Examples may include prescription medicines, phototherapy, over-the-counter products, and other therapies including non-drug therapies such as limiting exposure to sun, diet modification, etc.)
a.What specific symptoms do your therapies address?
b.How has your treatment regimen changed over time, and why?
5.How well does your current treatment regimen control your condition?
a.Would you define your condition today as being well managed?
6.Assuming there is no complete cure for your condition, what specific things would you look for in an ideal treatment for your condition?
a.What would you consider to be a meaningful improvement in your condition (for example specific symptom improvements) that a treatment could provide?
"Remember.Research is the key to your cure!"
AIP & Life story of Claire Sadoniczak
Wednesday - November 30, 2016 @ 09:00:24
Claire Sadowniczak
Type of Porphyria:
Acute Intermittent Porphyria (AIP)
Claire Sadowniczak of Orlando, Florida, is a member of the APF. She keeps the group encouraged and laughing. Her stories about her turtle Alamo are sheer delight and lessons in tenacity for all of us.
My mother and I have AIP; I started attacks at age 11. One thing that gives me great pleasure is rescuing a wild animal, nursing it back to health and releasing it back into the wild. One rescued 5" brown and black Florida mud turtle left me a present, an egg. It hatched on a freezing day, so I couldn't release it. The black hatchling was smaller than a dime, the shell still soft when I picked her up. I carried her in the palm of my hand as I was preparing a tank of gravel with a plastic sour cream lid as her "pond". We went to the pet store to try to find food small enough for her. They said I'd never keep her alive and she's now 11 years old. When I finally put her into her tank, she ran to the front glass begging me to pick her up again. She ignored my screaming Pomeranian who was jumping up and down in front of her tank. I named her Alamo for her courage in the face of danger.
Water turtles do not have salivary glands or pulmonary muscles to swallow air to breathe. After being out of the water awhile, her mouth dries out and her beak "squeaks". When this occurs, I put her back into her pond. Once I picked her out of her pond and put her on hubby's lap, so I could clean her tank. She doesn't like him so she began squeaking her beak when I left the room, begging to get off his lap!
She potty-trained herself, too. I kept a towel on my lap when I held her. She realized that I didn't like her mess and hasn't had an accident since she was six months old. She just squeaks her beak to ask to go back into her tank.
I feed her with a plastic spoon. Since she is in the snapping turtle family, I didn't want her to associate fingers with food. She'll swallow anything presented on a spoon, including medicine and will follow a spoon anywhere! She also loves TV and gets very involved. If a car explodes on a show, she'll open her mouth at the TV.
Once she had a respiratory infection; turtles can catch colds from people. The vet told me to add a heater and thermometer to her tank to keep the water at 80 degrees. She did not like either, popping the heater off its rubber suction cups and bashing it and the thermometer against the rocks till I removed them. It is HER home after all.
Alamo enjoys being an only child and will attack a mirror until it is removed. Although the breed is supposed to be "vicious", she is a sweety with me. She once saw a piece of shiny fuzz on my sweater that she wanted to eat. She very gently tried to get it with the side of her mouth so she would not accidentally bite me through the sweater.
I carry her around under the hem of my T-shirt or in a pocket for hours, and her little head comes out like a periscope to look around. She sits on my lap in the car. In the vet's office, Alamo watches the other animals from my lap and feels quite safe and content. I put her on the grass in the back yard for exercise, and she runs straight back to me. Definitely a lap turtle!
Alamo's Mother, Claire Sadowniczak Prior to my diagnosis of Acute Intermittent Porphyria, I underwent abdominal surgery with sodium pentothal, went into cardiac arrest and a three month major porphyria attack. Therefore, when I learned that I needed gallbladder surgery, I was very nervous.
I found a general surgeon, Dr. Cesar Cabascango, who is not only familiar with porphyria, but I was his fifth patient with porphyria. Normally, with the laproscopic gallbladder procedure, you have surgery the same day as admission and go home the following morning. Because of my porphyria, Dr. Cabascango had me admitted the day before surgery, opened a central line with three ports in my chest, and infused large doses of dextrose. He continued to infuse dextrose during surgery and for an additional two and a half days in the hospital after surgery. By faxing information on porphyria to my insurance company, he even got them to approve the additional stay in the hospital. I did not suffer a porphyria attack whatsoever.
The surgery was performed at Florida Hospital Orlando. The Assistant Director of Anesthesiology handled my case personally, and brought me through the procedure with no problems. Everyone at the hospital researched porphyria, read the brochures from the American Porphyria Foundation that I provided, questioned me about it, and treated me with such special care that it was my most positive hospital experience ever.
Many people with porphyria have horror stories about past medical care, including myself, but things are improving and the brochures provided by the American Porphyria Foundation are a great benefit when distributed to health care professionals.
Let's get Down on VP
Monday - November 28, 2016 @ 10:00:00
Let's get to know more about VP!
What is Variegate Porphyria?
VP is caused by a mutation in the enzyme protoporphyrinogen oxidase (PPOX), which is part of the pathway that produces porphyrins and heme. Acute attacks are similar to those in AIP and HCP but are unusual. A more common sign of the disease is blistering skin lesions, which are chronic in many people with VP.
Acute attacks almost always start with severe pain in the abdomen but sometimes in the chest, back, or thighs, and are often accompanied by nausea, vomiting, and constipation. Heart rate and blood pressure are commonly increased. These symptoms and signs are all due to the effects of the disease on the nervous system. Confusion, convulsions, and muscular weakness, due to impairment of the nerves controlling the muscles, may lead to paralysis. An acute attack usually lasts for days or weeks. Recovery from severe paralysis is generally slow.
Who gets Variegate Porphyria?
VP is especially common in South Africa in individuals of Dutch ancestry, where it has been estimated that 3 in 1,000 of the white population are affected. It is much less prevalent in other countries. Like AIP and HCP, it is an autosomal dominant disorder, meaning that a mutation is present in only one of the pair of PPOX genes.
What causes Variegate Porphyria?
As in HCP, acute attacks of VP are unusual except in the presence of environmental activating factors, such as drugs, hormones, and dietary changes.
How is Variegate Porphyria Diagnosed?
Urine ALA and PBG are increased during attacks, but as in HCP, these may increase less and decrease more rapidly than in AIP. Plasma porphyrins are frequently increased in VP, in contrast to AIP and HCP, and the plasma of VP patients displays a distinctive fluorescence peak, which is diagnostic. Fecal porphyrins are also elevated and are predominantly coproporphyrin III and protoporphyrin.
What are treatments for Variegate Porphyria?
Management and prevention are the same as in AIP and HCP. Blistering skin lesions are much more common than in HCP and are not readily treated. The only effective preventive measure is use of protective clothing.
What is the long-term outlook after an attack of Vairegate Porphyria?
The prognosis is usually good if the disease is recognized and treated promptly, before nerve damage develops. Although symptoms usually resolve after an attack, recovery of neuromuscular function (in a severe case) may require several months. Mental symptoms may occur during attacks but are not chronic. Premenstrual attacks often resolve quickly with the onset of menses.
Can attacks be prevented?
Yes, particularly with regard to drugs and diet. Genetic VP carriers should become informed on medications to avoid (see information on AIP and HCP) and should be prepared to point their healthcare providers to on-line drug lists that are regularly updated. The American Porphyria Foundation offers a mobile phone app that pulls up this information on line (http://porphyriadrugs.com/). A Medic Alert bracelet is useful for a situation in which the patient is incapacitated. Very frequent premenstrual attacks can be prevented by a gonadotropin-releasing hormone (GnRH) analogue (Lupron, Zoladex, others) administered with expert guidance. In selected cases, frequent noncyclic attacks can be prevented by once- or twice-weekly infusions of hemin.
Individuals who are prone to attacks should consume a normal balanced diet. Despite on-line discussion, there is no evidence that pushing carbohydrate prevents attacks, and it has the side effect of weight gain, which is undesirable for most people. Fasting, fad diets (for example, high protein) and gastric reduction surgery should be avoided. If weight loss is desired, it is advisable to consult a physician and a dietitian about an individualized diet with modest caloric restriction (ca. 10%), which will produce gradual weight loss without increasing the risk of an attack of porphyria. Exercise is safe in porphyria, and recommended.
EPP Fabian family takes on Orlando FL Vacation
Tuesday - November 15, 2016 @ 10:00:16
As many of you know living with EPP can be tricky. When deciding and preparing for our family trip. We knew it would be difficult but we choose to conquer together and have a wonderful time in the Orlando Florida area. How would we get through the Sun, was there any Shade? To our surprise with a few calls and planning we had a wonderful family vacation. Here are some things that we did to help make our trip enjoyable. Hope you all enjoy!
Both of my adult children, Kevin and Katie, have EPP. I never risked a trip to Orlando when they were younger due to cost and risk of them having a reaction to the sun, ruining a very expensive trip. Fortunately, we were able to go in November 2016. It was the best trip we have ever taken together as a family.
We stayed on Universal Studios property at the Hard Rock Hotel. This included 4 day passes to Universal with Fast Passes on every ride (except a couple in Harry Potter World). The Fast Passes WERE THE BEST EVER it allows you to go in a line separate from the regular line. Luckily, we went at a time the parks werent particularly busy. (I later learned that February is the least busy time. A driver also said Tuesdays and Thursdays are the least busy days of the week. Additionally, early afternoon is least busy as families go back to their hotels with young children to take naps).
We did present their doctors notes at the Customer Courtesy desk at Universal and were given one pass for our entire family to go ahead of any Fast Pass line that we needed. The young man did not even need to read the notes he was so accommodating and friendly. As it turns out, we did not need to present the medical pass as most of the lines were quick with the Fast Pass or in the shade and not necessary.
The first two days were CLOUDY (yeah!!!) though good quality sunscreen was applied regardless, as well as gloves, hats, long sleeve shirts and pants worn. Temps in the 70s was perfect. We spent our first two days at Universal.
Third day, VERY SUNNY, but we set this day to go to Hollywood Studios and the Magic Kingdom we took Uber and ventured to Hollywood Studios in the morning and afternoon. We stopped at Guest Services again, a VERY nice young man told us we really didnt need the doctors pass. We would just tell the worker in whatever line that we needed to go to the front. We had 3 Fast Passes that I scheduled our times in advance on the APP on my phone and it synced with our tickets. All 3 of these rides/events were inside. There were many shady areas, stores, and outdoor seating with shade. We werent there for kid rides and such and we planned exactly what we wanted to do for the most part. For us, we had no need to go to the head of any Fast Pass lane we were in.
We took the shuttle to the Magic Kingdom around 3 pm. I was able to schedule (with the APP) a Fast Pass one at a time. We chose the Haunted Mansion (it had a 2 hour wait) we were on the ride within 10 minutes. We saw a few other indoor events. As the sun was down by 5:30, we were free to roam around until the show started. I chose this day to go to Magic Kingdom since they were open later with a fireworks show at 9 pm. We got our spot to view the MOST SPECTACULAR fireworks display weve ever seen.
Our 4th day, Kevin spent his time in the Harry Potter parks while Katie and I went poolside. She was in the shade, and I was in the sun. We were on opposite sides of the pool, but later could find a mutually enjoyable spot. We went back to Universal around 5 until it closed at 8 pm. City Walk is open until 2 am with restaurants, and we walked over there for dinner OUTSIDE. J
Our last day, we went back to Universal using those Fast Passes for rides we missed, and then we spent a couple hours in the Wizarding World of Harry Potter. Those two parks are truly the shadiest place in the two Universal parks (especially Diagon Alley).
Staying protected with cover up and sunscreen, shadow hopping, taking advantage of the Fast Passes, and planning helped us have a SUN REACTION FREE vacation that weve always dreamed of in Orlando FL. P.S. Stay hydrated as well the parks allow you to bring in a bottle of water.
If you would like more tips on EPP & sun in the fun check out porphyriafoundation.org for all your needs including camps for the kids.
Porphyria News Save the Dates!
Thursday - November 10, 2016 @ 09:30:05
**ATTENTION ARIZONA PEOPLE**
We are happy to announce that a doctor trained through our Protect the Future program to train the next generation of experts has moved to Phoenix. Porphyria expert, Dr. Danielle Nance, who is board certified in Hematology and Internal Medicine, is in private practice and is now the Director of the Hemostasis and Thrombosis Clinic at the Arizona Bleeding Disorders Health and Wellness Center (Address: 821 N 5th Avenue, Phoenix, AZ 85003).
In addition to porphyria, this clinic also focuses on benign hematologic conditions, specifically hemophilia, bleeding and clotting disorders. The staff is very sensitive to the needs of chronic illness and high in-tensity medical conditions. They offer social services, wellness services, and have several great pharmacy and infusion service partners.
You can make an appointment with Dr. Nance by calling (602) 680-7722. If you are able to, we encourage all porphyria patients to see an expert to manage their care. We know you will enjoy meeting Dr. Nance!
You are invited to an American Porphyria Foundation Patient Education Meeting in San Diego, CA!!
Friday, December 2, 2016 5:30-8:30PM PST
* Presentation by A World Renowned Porphyria Expert * Opportunity to Participate in a Q & A Session * Meet Friends who Share Your Experiences with Porphyria at the Meet & Greet from 5:30-6:30PM * View the Latest Educational Material from the American Porphyria Fdn.
Seating is limited Please RSVP: 1-866-APF-3635 or Email: porphyrus@porphyriafoundation.com
Wyndham Garden San Diego 3737 Sports Arena Blvd. San Diego, CA 92110
APF Meeting Hosted by Evelyn Jacobucci Information
Wednesday - November 9, 2016 @ 10:30:17
APF member, Evelyn Jacobucci, will be hosting the next Patient Education Meeting on Friday, November 18, 2016 from 5:30-7:30 PM MST. You are welcome to bring your friends and family, but please be sure to RSVP to the APF or directly to Evelyn at 303-989-2073 or ejb2003@live.com.
Bear Valley Church 10001 W. Jewell Avenue Lakewood, CO 80232
*Presentation about Porphyria and Q & A Session *Meet Friends who Share Your Experiences with Porphyria *View the Latest Educational Material from the American Porphyria Foundation
ALL USA PORPHYRIA PATIENTS Enrollment for Longitudinal Study
Monday - November 7, 2016 @ 10:30:17
ALL USA Porphyria patients.
We need each of you to sign up for the studies of Porphyria. All types are able to join this study. The Porphyria Consortium are looking for volunteers now for the longitudinal study. Please read the following study opportunity: click on the below links to register and if you have any questions please contact the APF 1/866/APF/3635. If you have any questions please call the APF during normal business hours or leave a detailed message with a call back number and we will reach out to you.
{Here you can join the contact registry. Once you join the RDCRN Contact Registry, you will receive emails with information about current research studies for your disease. In these emails, you will be given information about how to join the research study. You will also be able to contact the researchers to ask any questions about the study.}
Please consider this study for all of those who suffer from Porphyria Disease. Please sign up and share this message with anyone who may benefit.
Gail Hubler & AIP Story
Thursday - November 3, 2016 @ 11:15:04
I am Gail Hubler and I have AIP (Acute Intermittent Porphyria). It was dormant until I had a tetanus shot at age 53. I had some issues with pain through out my life but thought it was from standing too long at work and from accidents in car and falls. When I got home from the hospital my entire body had changed and I was trying for seven years to find out WHY! I had every test know to man and beast but found nothing wrong, thus, it's stress from your job. So I quit my job which didn't help. One day my neighbor found an article in the newspaper in the section where you ask a Doctor questions. This man's symptoms were just like mine and so many weird ones I thought this has to be it. My Doctor said he has only had one patient with Porphyria and he was from Wales.
Most of my relations came from Wales. During an attack, I was tested and found out. I have battled how to keep myself from attacks for eight years now. I Have had to get off some of my medications and change some of them to another type so that they were safe to take with an Acute Porphyria. I have not had a drink of alcohol for 13 years. When I had a drink it would make me so sick, I knew it had to go. Before I was diagnosed I couldn't understand why previous I had a drink on weekends for years. I was always confused and could get lost in a card board boxonce I found out that was clear to me. So many things to watch out for and it's still a daily struggle to stay well. Tired all the time from anxiety. My worse attack came before I knew I had it and it was in the hospital after surgery! I had to educate myself on this disease and try to help others if I can. I swim and take medication and manage it to the best of my ability. Thank you Gail for sharing a brief glance of some of the changes you have made in your life to keep your AIP from flaring. For more tips from the Porphyria Experts please visit:www.porphyriafoundation.orgunder Acute Porphyrias
If you would like to share you story on your Porphyria please email me directly to Amy.APF@gmail.com "Remember.Research is the key to your cure!"
2nd Annual "Moo've It In The Moonlight" Run
Tuesday - November 1, 2016 @ 10:30:25
RSVP now for the 2nd Annual "Moo've It In The Moonlight" Run in Burlington, NC happening THIS SATURDAY at 6:00PM. APF member, Shawn Willis, will be hosting this event in the evening to highlight the challenges of those with light sensitivity. There will be a 5K AND a 1 Mile Untimed Fun Run and proceeds from the race will benefit the APF. Don't forget to RSVP online today!!
My name is Jason Barrett, and I was diagnosed with Erythropoietic Protoporphyria when I was 11 years old.
My story is similar to many others shared by those who live with EPP. I to have become intimately acquainted with the confusion, frustration, social isolation, and pain, as well as the undeserved feelings of guilt, depression, self-doubt, fear, and shame that are so common in the lives of those touched by this rare disorder.
I have carved a good, happy life for myself around the challenges that I face. I have beautiful young children who bring me joy and just accept that Daddy wears a hat. My wife trusts and believes in me, and together we purchased a wonderful home in a lovely neighborhood. I excel at my chosen profession in the electrical field despite the difficulties associated with being a working disabled person. I love cloudy or stormy weather and the twilight hours, and I use such time to enjoy the outdoors as much as I can.
When I had each of my children, I wondered what they would think of me and the difficulties I have faced. Would they know how strong I have had to be, or would they see me as a masked weirdo standing on the shady sidelines? It took a lot of thought, but I eventually decided to write about my experiences and perceptions in the hope that they might see the world through my eyes one day and understand.
Privately locking these things away for only my immediate family to someday read seemed somehow wrong. I remembered the fear I felt as a child as my family and I blundered through ignorance as though I was the first person ever to handle this type of situation. I remembered how lonely and alien I felt. I know that there are families still struggling through ignorance, who may feel that they are the first, who may feel alone and alien.
In September 2016, after I received a letter from the American Porphyria Foundation, I mustered up the courage to start a blog about being alive while experiencing EPP. I write about perceptions and experiences that I hope can be useful, uplifting, and insightful to my family, and to any stray reader who happens along.
EPP comes with sadness and pain, and sometimes those are the only things I feel. But there is also happiness and humor in my life. I want to share that living in darkness doesn't have to mean living in perpetual gloom. I am just weathering the storm of life as best I can like anyone else.
If you feel alone or alien because of your affiliation with EPP, know at least that youre not the first, just like I learned I wasn't the first, to have experienced this disorder. Even if you dont believe it now, you can create happiness and hope in your world. If there is anything EPP can teach us, it is that we all can learn to find joy and beauty in a storm.
Storms are in the eye of the beholder.
To view Jason's Blog about living and coping with EPP please visit: eppalive.wordpress.com.
To learn more specific things about EPP, FDA Patient meeting and more please visit: www.porphyriafoundation.org
"Remember.Research is the key to your cure!"
Clinuvel Information
Friday - October 21, 2016 @ 10:30:27
CLINUVEL PHARMACEUTICALS LTD[ASX: CUV; Nasdaq International Designation ADR: CLVLY; Xetra-DAX: UR9] today announced that it will meet with the US Food and Drug Administration (FDA) on 7 November to formally discuss lodging its new drug application (NDA) for the novel drug SCENESSE (afamelanotide 16mg). The pre-NDA meeting will focus on finalising requirements for filing SCENESSE with the FDA for the treatment of adult patients with the rare genetic disorder erythropoietic protoporphyria (EPP).
This is the last step required of CLINUVEL before we ask the FDA to review the risk-benefit profile of SCENESSE in a submitted dossier, CLINUVELs Acting Chief Scientific Officer, Dr Dennis Wright said. A successful risk-benefit review will enable CLINUVEL to make the drug available to US EPP patients.
The pre-NDA meeting, with the FDAs Division of Dermatology and Dental Products (DDDP), is part of ongoing dialogue between CLINUVEL and the FDA since CLINUVEL commenced a clinical program for EPP in 2006.
Earlier this year the FDA granted SCENESSE Fast Track designation, enabling among other benefits a rolling review of the NDA dossier. The FDA also completed an initial review of CLINUVELs clinical data package, deeming it satisfactory and sufficient for NDA submission. On 24 October the DDDP will host an EPP Workshop to obtain the patients and physicians perspective on certain disease areas, including the effectiveness of treatments.
SCENESSE is the first treatment ever evaluated in contemporary clinical trials for EPP. The drug provides photoprotection to EPP patients who suffer from acute reactions to visible light and sun (phototoxicity). In 2014 SCENESSE was approved in Europe for the prevention of phototoxicity in adult EPP patients, with the drug now prescribed in a number of European countries.
It has always been our goal to make the drug available to US EPP patients, and it has become more pressing now that the drug is available in Europe, CLINUVELs Global Director, Regulatory Affairs, Ms Nicoletta Muner said. In recent months our dialogue with the FDA has intensified as the agency sought to learn more about our program and as the product is being distributed internationally. We look forward to the next steps in the review of SCENESSE.
"Remember.Research is the key to your cure!"
Accused of Being a Hypochondriac, Lisa is Finally Diagnosed with Acute Intermittent Porphyria- Global Genes
Wednesday - October 19, 2016 @ 13:10:24
Accused of Being a Hypochondriac, Lisa is Finally Diagnosed with Acute Intermittent Porphyria
A swollen stomach made her look pregnant, but the pain burning, shocking, horrible, out-of-this world, as she describes it was worse than childbirth, leaving her doubled over and gasping for breath. Symptoms came and went, seemingly at random. Vomiting. Constipation. High blood pressure. A racing pulse. Numbness in her hands. Paralysis in her right foot.
Lisa Kehrberg spent several days in a hospital, but every test came back normal. And the doctors began to wonder if perhaps she had been under a lot of stress lately.
As a physician herself a specialist in pain management, as a matter of fact Kehrberg knew how to translate that question: Theres really nothing wrong with you.
Actually, no, she told the doctors. She hadnt been under stress. Im really sick right now. Can you help me?
You need to calm down, one health-care worker told her. Go home. Take it easy. And pull yourself together.
Kehrberg had been sick before. The first bout came 22 years ago, when she was going to high school in Bartlesville. Another one hit during college, when she was briefly hospitalized with abdominal pain. But it wasnt until September 2013, while she was working as a doctor for U.S. Department of Veterans Affairs in Chicago, that Kehrberg finally demanded answers.
Released from one Chicago hospital without a diagnosis, she checked into another one. Did the doctors there think she was a hypochondriac?
Yeah, Im sure they did, she said. Doctors are too quick to give up. Its stress. Its your imagination. Its not as bad as youre saying it is. They dont want to believe that theres something wrong with you if they dont know what it is.
The second hospital did all the usual tests, with the same results. Normal. Why not try something different?
Its almost a cultural thing for doctors, she said. Its looked down upon to start testing for rare diseases. Nobody wants to look like theyre grasping for answers. It cant be that, so it must be your imagination.
Kehrberg remained undiagnosed until a hospital nurse noticed that her urine had turned dark brown, a classic symptom of a rare genetic disorder called Acute Intermittent Porphyria, by some estimates afflicting less than 0.0001 percent of the population.
Kehrberg had heard of it herself but never considered the possibility. While not curable, the disease can be controlled to some extent by diet and medication.
More than a year later, Kehrberg now speaks as an advocate for people with Porphyria and other rare conditions. And she came back to Oklahoma recently for a fundraiser at her parents ranch near Pawhuska.
Her message to patients: Speak up. Trust your instincts. Demand answers.
And to physicians: Listen to people and believe what they are telling you. There arent a lot of hypochondriacs walking around out there. There are a lot of sick people who need help.
Important Things To Know Before The FDA Meeting For EPP On MONDAY
Monday - October 17, 2016 @ 13:39:20
Important Things To Know Before The FDA Meeting For EPP On MONDAY
The FDA Meeting for EPP is just around the corner! The FDA has distributed additional information for all registered attendees. The check in process is lengthy, so everyone needs to be sure to arrive to the FDA at 9AM on Monday, October 24th. As a reminder, the APF will have a shuttle bus available to transport attendees between the Holiday Inn College Park and the FDA.
Transportation: If you are taking a cab, ask the driver to drop you off at Building 1. If you will be driving and need to park, there will be signs labeled Event Parking pointing you in the direction of various surface parking lots (Southeast, 132A and 132B). Parking spots may be difficult to secure that will be close to Building 1, but there will be shuttles circulating to all parking lot bus stops every 5 minutes that can pick you up. Make sure to tell the shuttle driver to drop you off at Building 1. Here is FDAs transportation page for more information: http://www.fda.gov/â?¦/WhiteOakCampusInformation/ucm241740.htm
Arriving: Please make sure to first arrive at Building 1 in order to clear security and enter FDA as a guest. Building 1 is the first building you see when you arrive on campus, behind the circle with flags. You will need to present a government issued ID. Once in Building 1, visitors will have to go through the x-ray and magnetometer machines and then go down a corridor to the Building 31 Great Room. The meeting will be taking place here in the Great Room, or Rooms 1503 B and C. There is a small portion of the path between Building 1 and 31 where visitors will be outside. There are stairs and a ramp between the buildings. We will have FDA staff helping direct visitors to the meeting room.
Registration: Please plan to arrive between 9:00-9:45am. We will have sign-in sheets available on the tables inside the room. Luggage can be kept in the meeting room, either with the individual or in the back of the room. Sara and Meghana will both be in the room to help you with anything else you might need. If you have any difficulties finding the meeting room, or if there are any immediate questions you may need to ask, Meghanas cell phone number is 772-342-1816, and Saras is 412-606-9445.
Lunch: There is a kiosk in the foyer of the meeting room where food and beverages are available for purchase. You can pre-order your lunch in the morning before the start of the workshop; if you choose to pre-order, your lunch will be available for pick-up inside the meeting room itself (where we are planning to keep the lights dimmed) during the lunch break.
Example of Past Meetings: We have conducted meetings similar to this one in the past. Please find a link to a similar meeting we held for psoriasis here: http://www.fda.gov/â?¦/â?¦/PrescriptionDrugUserFee/ucm470608.htm. Here you will find the webcast recording and full transcript of the meeting.
Agenda: The workshop agenda is now available on the workshop webpage: http://www.fda.gov/Drugs/NewsEvents/ucm501389.htm. The patient panel will kick off the discussion in the morning, and will be followed by a large-facilitated discussion with patients and caregivers from the audience. So we encourage you to share your experience during the large-facilitated discussion portion of the workshop. The workshop is in-tended to be an opportunity for FDA to hear directly from patients, and we make every effort to make sure the voice of the patient is heard. We also encourage you to submit your comments directly to our public docket. This is a website through which patients living with EPP and oth-ers can share their experiences and perspectives. These comments supple-ment the input we get from the workshop. We will review all of these com-ments. To submit your comment, visit: https://www.regulations.gov/document?D=FDA-2016-N-1493-0001. Click on the Comment Now button in the top-right corner. You will be able to upload a PDF or Word version of your comments.
If you have any questions, you can contact the FDA at megha-na.chalasani@fda.hhs.gov. We are all looking forward to this workshop!
The FDA Meeting for EPP is Less Than 3 Weeks Away!
Friday - October 14, 2016 @ 10:30:05
The FDA Meeting for EPP is Less Than 3 Weeks Away!
The APF is busy preparing for the upcoming EPP meeting at the FDA on October 24th, 2016. We are very excited about the large number of attendees and hope even more people RSVP to us in the coming weeks.
When registering for the FDA meeting, there is an option to indicate if you are interested in participating in a panel. If you have signed up to participate in the panel during the meeting, you should have received an email with a list of discussion questions from Meghana Chalasani (FDA meeting coordinator), in addition to your registration confirmation email. These questions must be answered and returned to Ms. Chalasani.
The deadline to submit your answers for the panel has expired.
You will be notified of your panel status at least 1 week in advance of the meeting. If you have signed up to speak at the panel, but did not receive the follow up email from Ms. Chalasani, then you are not being considered for the panel. You must contact Ms. Chalasani to receive the questions.
**PLEASE let the APF know if you are selected for the panel. If you are still unsure whether or not you can attend this important meeting, feel free to contact Jessica@porphyriafoundation.org with any questions/comments you have. We are excited to meet everyone in a few short weeks!
As a reminder, the deadline to register for the FDA Meeting in person OR via webcast is Monday, October 17th, 2016!
If you are not registered, you cannot attend the meeting, so be sure to register ASAP! Visit this website for registration:
Taking Notes and Recording Your Visit~ Have you ever felt overwhelmed during a Doctor's appointment in which you received a large amount of information? I know I have. Perhaps you didn't ask any questions at the time even though you felt you had questions once you had time to process all of the details. Your not alone. Many patients find themselves with many unanswered questions once they have a chance to look over the printed materials and digest the information. These tips may help you feel more informed during your next visit:
Write down a list of specific questions.
Bring someone to the appointment with you to listen, take notes, and record the visit.
Verbally summarize the instructions for the doctor or the nurse
Ask pointed questions such as: "can you please review with me again what I need to know about getting Porphyria testing done right?"
Many health care providers are using a technique called "Teach-Back" to ensure you understand what they just explained. They may ask you open ended questions rather than just expecting a yes or a no response. This is an opportunity for you to recap what you just heard which helps to reinforce the information. Have fun trying this new technique at your next Doctor's appointment! "Remember.Research is the key to your cure!"
Your Stories
Wednesday - October 5, 2016 @ 13:01:33
YOUR STORIES- Are you a porphyria survivor or know someone with an inspiring story to tell? How has porphyria affected you or your loved ones?
Whether it's a story, a poem, a picture, fundraising event, a picture or a painting, share YOUR story in your way.
Your story will be made available in a published blog and the upcoming 2017 Porphyria Awareness Week. Please email me your type & name with what you want to share Amy.apf@gmail.com
A journey to successful protection with Scenesse. #SuccessWithScenesse
Thursday - September 29, 2016 @ 14:20:45
A journey to successful protection with Scenesse. #SuccessWithScenesse
Our son, JT von Seggern has Erythropoietic Protoporphyria, EPP. EPP negatively effects the liver, results in vitamin D deficiencies and causes excruciating pain all from exposure to the sun. JT has experienced the painful effects of EPP since he was 2 years old. It took us 9 years of extensive research and numerous doctors visits, including a trip to Johns Hopkins to finally get a diagnosis for what was causing our child so much pain. While gaining a diagnosis was a huge relief, learning that there were no treatments and no cure for EPP quickly knocked the air out of our initial relief. Exactly how does one go about protecting their now 11 year old, sports loving son, from exposure to the sun? Daunting. Through the ensuing years, JT experienced frequent pain, swelling, scarring, missed school days, and numerous rounds of Prednisone to bring down the swelling caused from exposure to the sun. Flash forward to the spring, 2015. The years of dealing with EPP and the understandable anxiety that accompanies such a cruel disease started to really take a toll on JT. By the time we picked him up from college at the end of the spring semester, it was agonizingly apparent that this cycle could not continue. In May, 2015, we asked Dr. Silverman, the doctor treating JT if there were any new developments in treating EPP. There were none in the U.S. but Dr. Silverman mentioned a promising drug containing afamelanotide. I indicated that I wanted JT on that drug and while Dr. Silverman was sympathetic to JTs plight, he informed us that it would be next to impossible to get access to the drug for JT. At that point, the mama bear in me went into full fight mode to protect. Through a miraculous array of previous untraveled avenues, a happenstance meeting in New York, calls to Australia, Italy the U.K. and finally a contact in Zurich. 12, 670 miles traveled 2 cab rides 2 Trams 1 hotel 1 hospital 1 implant 2-3 months of vital protection JT received his first implant of Scenesse in Zurich, Switzerland on August 11, 2015. The protection that JT received from this first implant of Scenesse was nothing short of miraculous and life changing. A whole new world has opened up for JT. He only missed one day of classes due to over exposure, his grades went up across the board, and he gained back his confidence and left anxiety in the dust of his shadow. â?¦a shadow that was made possible by the successful protection from Scenesse. The journey to successful protection with Scenesse is not an easy one, but it is the only option available until the FDA accelerates approval for afamelanotide 16 mg. As we pulled up to Dulles Airport for JTs second implant scheduled for January 12, 2016, a beautiful rainbow greeted us and we knew we continued to be on the right path. JT was all smiles as we awaited our flight to Zurich We arrived in Zurich at 7:55 am on Monday the 11th. JT rested up and prepared himself mentally for the procedure. The hospital is just up the hill from the Tram station and off we trekked to Pavillion F in the hospital where the implant would take place. We were MUCH better at navigating this adventure since we had done this during our first trip in August. Tuesday morning we arose early, ate and walked a few blocks to the closest Tram station and took the #14 to Triemli Hospital After we arrived at the designated spot, we waited for JT to be called into the procedure room. The numbing patch that Dr. Minder had given me in August to apply to JTs side did not totally numb him so after cleaning his side with iodine, Dr. Minder gave him an injection to thoroughly numb the area. JT was called into the procedure area where we met with Dr. Minder and her assistant. Dr. Minder went over the procedure and then she prepped JT for the implant. Next up, the actual implant! After it was all done and JT was bandaged up, JT rested for a bit just to make sure he was fine to make the trek back to the hotel. Knowing Scenesse was in his body and would soon be protecting him from the sun made even this rainy Tuesday one full of hope and promise. Singing in the rain was not out of the question! ï?? We walked back to the #14 Tram for the 20-minute ride back to the stop near our hotel. JT rested the rest of the day. Dr. Minder, Rocco Falchetto and Jasmin Barman joined us for a send off dinner. Both Rocco Falchetto and Jasmin Barman are EPP patients as well and being successfully treated with Scenesse. After a good nights rest, we left our hotel and boarded our flight back to Virginia. 12, 670 miles traveled 2 cab rides 2 Trams 1 hotel 1 hospital 1 implant 2-3 months of vital protection 12, 670 miles traveled 2 cab rides 2 Trams 1 hotel 1 hospital 1 implant 2-3 months of vital protection JTs confidence knowing he is protected from pain for another 2 months shows on his face! My question is, why? Why must we as American citizens go to such lengths to provide successful, safe protection from the sun? Accelerated approval for Scenesee (afamelanotide 16mg) is needed NOW. No EPP patient should be needlessly suffering. FDA, Approve Scenesse All this will be repeated again on March 8 for JTs 3rd implant of Scenesse. The lengths to which one will go through to protect their child are incredible. And no EPP patient should have to endure all of this to live a pain free, healthy life.
Do you know about EPP or XLP
Tuesday - September 27, 2016 @ 10:38:21
Erythropoietic Protoporphyria (EPP) and X-Linked Protoporphyria (XLP)
Erythropoietic Protoporphyria (EPP) or Protoporphyria
Erythropoietic Protoporphyria is characterized by abnormally elevated levels of protoporphyrin IX in erythrocytes (red blood cells) and plasma (the fluid portion of circulating blood), and by sensitivity to visible light that is usually noticed in early childhood and occurs throughout life. EPP can result either from mutations of the ferrochelatase gene (FECH), or less commonly the delta-aminolevulinic acid synthase-2 gene (ALAS2). When EPP is due to an ALAS2 mutation it is termed X-linked protoporphyria (XLP), because that gene is found on the X chromosome.
Protoporphyrin accumulates first in the bone marrow in EPP, and then in red blood cells, plasma and sometimes the liver. Protoporphyrin is excreted by the liver into the bile, after which it enters the intestine and is excreted in the feces. It is not soluble in water so is not excreted in the urine.
EPP is the third most common type of porphyria, and the most common in childhood. It causes very painful photosensitivity and can greatly impair quality of life. Delay in diagnosis is greater than with any other type of porphyria.
Swelling, burning, itching, and redness of the skin may appear during or after exposure to sunlight, including sunlight that passes through window glass. This can cause mild to severe burning pain on sun-exposed areas of the skin. Usually, these symptoms subside in 12 to 24 hours and heal without significant scarring. Blistering and scarring are characteristic of other types of cutaneous porphyria but are unusual in EPP. Skin manifestations generally begin early childhood and are more severe in the summer.
There is an increased risk of gallstones, which contain protoporphyrin. Excess protoporphyrin can also cause liver damage. Less than 5% of EPP patients severe liver damage and a condition caused protoporphyric hepatopathy that sometimes requires liver transplantation.
Diagnosis and Genetic Counseling
EPP should be suspected in anyone with non-blistering photosensitivity especially when it is prolonged and beginning in childhood. It is easy to make a diagnosis, or rule it out, once it is suspected.
The diagnosis of EPP is established by finding an abnormally high level of total erythrocyte protoporphyrin, and showing that this increase is mostly free protoporphyrin rather than zinc protoporphyrin. There is considerable confusion about which test to order. Sometimes laboratories have measured only zinc protoporphyrin and reported results incorrectly as protoporphyrin or free erythrocyte protoporphyrin (FEP). Laboratories that measure total erythrocyte protoporphyrin, free protoporphyrin and zinc protoporphyrin and report results reliably are:
Porphyria Laboratory and Center, University of Texas Medical Branch at Galveston, 1-409-772-4661
Mayo Medical Laboratories, 1-800-533-1710
ARUP Laboratories
Porphyrins are almost always elevated in plasma in EPP, but may be normal in mild cases. Fecal porphyrins may be normal or increased.
An experienced biochemical laboratory can usually distinguish between patients with EPP and XLP, because the former have much less zinc protoporphyrin in their erythrocytes. This can be explained because in the marrow the enzyme ferrochelatase not only normally makes heme (iron protoporphyrin) from protoporphyrin and iron, but can also make zinc protoporphyrin, especially when excess protoporphyrin is present or iron is deficient. However, this does not replace DNA studies.
Rarely, EPP develops in adults in the presence of a bone marrow disorder such as polycythemia vera, and is due to expansion of a clone of red blood cell precursors in the marrow that is deficient in ferrochelase.
DNA studies are important for confirming the diagnosis of EPP and XLP and for genetic counseling. This should be completed first in a person known to have the disease, and the information about the mutations in that individual used to guide testing of family members.
When EPP is due to a FECH mutation the inheritance is described as autosomal recessive. It is most common to find that one severe mutation is inherited from one parent and another weak mutation inherited from the other parent. The weak mutation is quite common in normal Caucasians, rare in Blacks and even more common in Japanese and Chinese populations. This mutation is sometime referred to as hypomorphic because it results in formation of a less than normal amount of ferrochelatase. But is does not cause EPP unless it is paired with a severe mutation. The severe mutation is characteristic for an EPP family and is present in all affected individuals. Carriers of the severe mutation are not affected because they do not have the weak mutation. Affected individuals and unaffected carriers can transmit the severe mutation to the next generation. Some of their children will have EPP if the other parent has a copy of the weak mutation. Rarely, the weak mutation is absent in an EPP family and two severe mutations are found, with at least one producing some ferrochelatase.
In XLP, mutations of the ALAS2 gene, which is found on the X chromosome, causes an increase in the production of the enzyme ALAS2 in the bone marrow. Several of these gain of function mutations have been described in different XLP families. In XLP protoporphyrin production exceeds that needed for heme and hemoglobin formation. Like hemophilia and other X linked genetic diseases, XLP is more common in men. Women have two X chromosomes and are usually not affected because they have a normal as well as a mutated ALAS2 gene. Men have only one X chromosome and will be affected if they inherit an ALAS2 mutation. Women with an ALAS2mutation will, on average, pass that mutation to half of their daughters (who will usually be unaffected carriers) and to half of their sons (who will be affected).
Treatment and Management
1. Sunlight protection
Protection from sunlight is the mainstay of management of EPP, and this is necessary throughout life. Disease severity and porphyrin levels in erythrocytes and plasma probably remain high and relatively constant throughout life in EPP. However, this has been little studied and more longitudinal observations are needed. Life style, employment, travel and recreation require adjustment in order to avoid painful reactions to sunlight and even from exposure to fluorescent lighting. For these reasons EPP can substantially affect quality of life.
Protective clothing, including broad-brimmed hats, long sleeves, gloves and trousers (rather than shorts), is beneficial. Several manufacturers specialize on clothing made of closely woven fabrics for people with photosensitivity.
2. Beta-Carotene (Lumitene Tishcon)
Beta-carotene is an over the counter product that was originally developed in a purified form as a drug for the treatment of EPP, and was shown to be effective by Dr. Micheline Mathews-Roth at Harvard University and others. The pharmaceutical grade formulation is now distributed by Tishcon as Lumitene, and can be ordered by calling 1-800-866-0978 or via the website www.epic4health.com. Other products are less standardized and reliable and are not recommended.
Beta-carotene provides protection by quenching reactive oxygen products that form when protoporphyrin is activated in the skin by light. It is important to take an amount that is adequate to be protective. For more information about Lumitene, including a recommended dosing schedule, please see the Lumitene section of this website.
3. Other considerations
In an occasional patient, protoporphyrin causes liver problems, so monitoring liver function is important. EPP patients should also not use any drug or anesthetic which causes cholestasis (slowing down bile flow), and should also avoid alcohol. Women should avoid medications containing estrogen (birth-control pills, hormone replacement therapy), and men should avoid testosterone supplements, as these substances can also have deleterious effects on the liver of a person with EPP.
Consult a specialist. Because EPP is a rare condition, most physicians are not knowledgeable about it. Contact The American Porphyria Foundation, 713-266-9617 for contact with an expert and to provide further information. A Medic Alert bracelet with instructions to contact a specialist if needed is a worthwhile precaution.
Yearly monitoring. Testing to include erythrocyte total protoporphyrin, plasma porphyrin, complete blood counts, ferritin and liver function tests should be done yearly. Porphyry levels are expected to be stable and liver tests to remain normal. EPP patients may have evidence of iron deficiency, and an iron supplement may be advisable if the serum ferritin is below about 20 ng/mL.
Vitamin D. Because they avoid sunlight, EPP patients are likely to be deficient in vitamin D. A vitamin D supplement with calcium is recommended for bone health.
Liver protection. It is important to avoid other causes of liver disease that might promote the development of liver complications from EPP. Patients should avoid alcohol and other substances that might damage the liver, including many herbal preparations, and be vaccinated for hepatitis A and B.
Surgical lights. Strong operating room lights can cause photosensitivity of the skin and even surfaces of internal organs. Flexible membrane filters, such as CL5-200-X from Madico Co., are available to cover surgical lights and offer some protection. This is especially important in EPP patients with liver failure, which causes even greater increases in protoporphyrin levels and photosensitivity.
Drugs. Drugs that are harmful in other porphyrias are not known to make EPP worse, but are best avoided as a precaution. This may include estrogens and other drugs that might reduce bile formation. A short course of a non-steroidal anti-inflammatory drug can provide some pain relief after an episode of photosensitivity, but can cause ulcerations of the digestive track especially with prolonged use.
Laser treatment. According to Dr. Roth, laser treatments for hair removal or eye surgery have not been a problem in EPP people. But the doctor should be made aware of the diagnosis, and that laser output between 400 and 650 nanometers might be harmful. Before hair removal treatment, the doctor may irradiate a small area of the skin to be treated for the length of time it will take to do the hair removal to ascertain if the patient would react within the period of time that a reaction to sunlight would be expected in that patient.
Children with EPP. Avoiding sunlight can be difficult for children with EPP who have less sunlight tolerance than their friends. Camp Discovery is an option for such children. It provides a week-long summer camping experience of fishing, boating, swimming, water skiing, arts and crafts, and just plain fun for young people with skin disorders, and is sponsored by the American Academy of Dermatology. Full scholarships, including transportation, are provided by the American Academy of Dermatology through generous donations of their members and other organizations. Members of the Academy are asked to recommend candidates for Camp Discovery, so ask your child's doctor about sending your child to Camp Discovery.
Clinuvel Pharmaceuticals is developing afamelanotide (Scenesse) for the treatment of EPP. This drug is given by injection and increases skin pigmentation. Another study of this drug is expected to open within the next year.
All patients with porphyria are encouraged to enter the Porphyrias Registry at the Porphyrias Consortium website. A link to this website is found on the website of the American Porphyria Foundation. Registration demonstrates to NIH that patients and their families think that research on porphyrias is important. You can also ask that one of the 6 porphyria center in the Consortium contact you.
Additional Reading about EPP and below that is more info on XLP:
Erythropoietic Protoporphyria
NORD gratefully acknowledges Micheline M. Mathews-Roth, MD, Associate Professor of Medicine, Harvard Medical School, for assistance in the preparation of this report.
Synonyms of Erythropoietic Protoporphyria
EPP
Erythrohepatic Protoporphyria
Protoporphyria
General Discussion
Erythropoietic protoporphyria (EPP) is a rare inherited metabolic disorder characterized by a deficiency of the enzyme ferrochelatase (FECH). Due to abnormally low levels of this enzyme, excessive amounts of protoporphyrin accumulate in the bone marrow, blood plasma, and red blood cells. The major symptom of this disorder is hypersensitivity of the skin to sunlight and some types of artificial light, such as fluorescent lights (photosensitivity). After exposure to light, the skin may become itchy and red. Affected individuals may also experience a burning sensation on their skin. The hands, arms, and face are the most commonly affected areas. Some people with erythropoietic protoporphyria may also have complications related to liver and gallbladder function. Erythropoietic protoporphyria is inherited as an autosomal dominant genetic trait with poor penetrance.
Erythropoietic protoporphyria is one of a group of disorders known as the porphyrias. The porphyrias are all characterized by abnormally high levels of particular chemicals (porphyrins) in the body due to deficiencies of certain enzymes essential to the synthesis of hemoglobin. There are at least seven types of porphyria. The symptoms associated with the various types of porphyria differ, depending upon the specific enzyme that is deficient. It is important to note that people who have one type of porphyria do not develop any of the other types.
Signs & Symptoms
The most common symptom of erythropoietic protoporphyria is hypersensitivity of the skin to sunlight and some types of artificial light (photosensitivity), with pain, itching, and/or burning of the skin occurring after exposure to sunlight and occasionally to fluorescent light. Affected individuals may also exhibit abnormal accumulations of body fluid under affected areas (edema) and/or persistent redness or inflammation of the skin (erythema). In rare cases, affected areas of the skin may develop sac-like lesions (vesicles or bullae), scar, and/or become discolored (hyperpigmentation) if exposure to sunlight is prolonged. However, scarring and/or discoloring of the skin is uncommon and rarely severe. These affected areas of skin may become abnormally thick. In addition, in some cases, affected individuals may also exhibit malformations of the nails. The severity and degree of photosensitivity is different from case to case. Photosensitivity is often seen during infancy; however, in some cases, it may not occur until adolescence or adulthood.
In some affected individuals, the flow of bile through the gallbladder and bile ducts (biliary system) may be interrupted (cholestasis) causing gallstones (cholelithiasis) to form. In turn, such stones can cause obstruction and/or inflammation of the gallbladder (cholecystitis). Rarely, affected individuals may also develop liver damage that, in very severe cases, may lead to liver failure requiring transplantation.
Symptoms usually start in childhood but diagnosis is often delayed since blistering is not common and, because the porphyrins are insoluble, they usually escape detection on urinanalysis. The diagnosis is made upon finding increased levels of the protoporphyrin in the plasma or red blood cells.
Causes
Erythropoietic protoporphyria is a rare disorder inherited as an autosomal dominant genetic trait with poor penetrance. Human traits, including the classic genetic diseases, are the product of the interaction of two genes, one received from the father and one from the mother.
In dominant disorders, a single copy of the disease gene (received from either the mother or father) will be expressed dominating the other normal gene and resulting in the appearance of the disease. The risk of transmitting the disorder from affected parent to offspring is 50 percent for each pregnancy regardless of the sex of the resulting child. The risk is the same for each pregnancy.
The symptoms of erythropoietic protoporphyria develop due to excessive levels of a chemical called protoporphyrin that accumulates in certain tissues of the body (i.e., the plasma, red blood cells, and the liver). Excessive protoporphyrin levels occur as the result of abnormally low levels of the enzyme ferrochelatase (FECH).
There are several different allelic variants of erythropoietic protoporphyria. An allele is any of a series of two or more genes that may occupy the same position (locus) on a specific chromosome. Symptoms of these allelic variants of erythropoietic protoporphyria are predominantly the same; however, one type may be inherited as an autosomal recessive genetic trait.
The gene that is responsible for regulating the production of the enzyme ferrochelatase (FECH) has been located on the long arm of chromosome 18 (18q21.3). Chromosomes are found in the nucleus of all body cells. They carry the genetic characteristics of each individual. Pairs of human chromosomes are numbered from 1 through 22, with an unequal 23rd pair of X and Y chromosomes for males, and two X chromosomes for females. Each chromosome has a short arm designated as p and a long arm identified by the letter q.
Some people who have inherited this defective gene may have slightly elevated levels of protoporphyrin in the body but will not exhibit the symptoms of erythropoietic protoporphyria.
Affected Populations
Erythropoietic protoporphyria is a very rare inherited disorder that affects males and females in equal numbers. It is estimated that the disorder occurs in about 1 in about 74,300 individuals. The onset of symptoms affecting the skin usually occurs in infancy; however, in some cases, onset may not occur until adolescence or adulthood. More than 300 cases of EPP have been reported in the medical literature.
Related Disorders
Symptoms of the following disorders can be similar to those of EPP. Comparisons may be useful for a differential diagnosis:
There are several other types of porphyrias, all of which involve deficiencies of specific enzymes. Most of the symptoms of these porphyrias are not similar to the symptoms found in erythropoietic protoporphyria. Individuals with porphyria cutanea tarda and congenital erythropoietic porphyria may develop skin lesions; however, these lesions do not resemble the skin lesions found in EPP. It is important to note that individuals with one type of porphyria do not develop any of the other types. In addition, there are skin disorders characterized by hypersensitivity to artificial light and sunlight besides EPP, such as xeroderma pigmentosum and epidermolysis bullosa. The skin lesions in these disorders do not resemble the skin lesions in EPP. (For more information on these disorders, choose Porphyria and Epidermolysis Bullosa as your search terms in the Rare Disease Database.)
Xeroderma pigmentosum (XP) is a group of rare inherited skin disorders characterized by hypersensitivity of sunlight and some types of artificial light, with skin blistering occurring after such exposure. In some cases, pain and blistering may occur immediately after contact with sunlight or artificial light. Acute sunburn and persistent redness or inflammation of the skin (erythema) are also early symptoms of xeroderma pigmentosum. In most cases, these symptoms may be apparent immediately after birth or occur within the next three years. Other skin symptoms of xeroderma pigmentosum may include discolorations of the skin, weak and fragile skin, and/or scarring of the skin. Xeroderma pigmentosum also affects the eyes; the most common symptom being an extreme intolerance to light (photophobia). Additional symptoms may include some neurological impairments, short stature, an increased susceptibility to some forms of cancer (e.g., skin cancer). There are several types of xeroderma pigmentosum; in most cases, XP is inherited as an autosomal recessive genetic trait. (For more information on this disorder, choose Xeroderma Pigmentosum as your search terms in the Rare Disease Database).
Diagnosis
The diagnosis of erythropoietic protoporphyria (EPP) may be made by a thorough clinical evaluation, characteristic physical findings, and specialized laboratory tests. EPP is usually diagnosed during infancy or early childhood, due to characteristic skin symptoms. The diagnosis may be confirmed by testing the red blood cells (erythrocytes) for increased levels of protoporphyrin.
Standard Therapies
Treatment
Avoidance of sunlight will be of benefit to individuals with erythropoietic protoporphyria. The use of topical sunscreens, double layers of clothing, long sleeves, hats, and sunglasses will also benefit photosensitive individuals. Individuals with EPP may also benefit from window tinting or using vinyls or films to cover the windows in their car or house. Before tinting or shading car windows, affected individuals should check with their local Registry of Motor Vehicles to ensure that such measures do not violate any local codes.
In erythropoietic protoporphyria, a high potency form of oral beta-carotene (Lumitene, Tishcon) may be given to improve an affected individual's tolerance of sunlight. For more information on this treatment, contact the organizations listed at the end of this report (i.e., American Porphyria Foundation and the EPPREF) and Mr. George McShane of the Tishcon Corp. (1-800-848-8442). In some cases, the drug cholestyramine may be given to alleviate skin symptoms and lower the protoporphyrin levels in the body.
When iron deficiency is present, iron supplements may be given. A type of bile acid (chenodeoxycholic acid) may be prescribed to help the liver dispose of excess protoporphyrin, and activated charcoal or cholestyramine may be used to interrupt the circulation of protoporphyrin through the liver and intestines.
Estrogens and drugs that can impair bile flow should be given cautiously under the supervision of a physician. In addition, individuals with high levels of protoporphyrin in the plasma and red blood cells should be observed closely by a physician for possible liver malfunction that could eventually lead to liver failure.
Genetic counseling will be of benefit for affected individuals and their families. Other treatment is symptomatic and supportive.
Investigational Therapies
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. Government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
The orphan product L-Cysteine is being tested for the prevention and reduction of photosensitivity in erythropoietic protoporphyria. More research is needed to determine the long-term safety and effectiveness of this drug for the treatment EPP. For more information, contact:
Red blood cell transfusions have also been used to treat some people with EPP. In some affected individuals with severe liver disease, liver transplantations have been performed. Extreme caution should be used by physicians considering these treatment options; each particular case should be evaluated on its own merits.
Hereditary Coproporphyria. (HCP) Porphyria Expert Dr. Bissell
GeneReviews[Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016. 2012 Dec 13 [updated 2015 Jul 1].
Excerpt
CLINICAL CHARACTERISTICS:
Hereditary coproporphyria (HCP) is an acute (hepatic) porphyria in which the acute symptoms are neurovisceral and occur in discrete episodes. Attacks typically start in the abdomen with low-grade pain that slowly increases over a period of days (not hours) with nausea progressing to vomiting. In some individuals, the pain is predominantly in the back or extremities. When an acute attack is untreated, a motor neuropathy may develop over a period of days or a few weeks. The neuropathy first appears as weakness proximally in the arms and legs, then progresses distally to involve the hands and feet. Some individuals experience respiratory insufficiency due to loss of innervation of the diaphragm and muscles of respiration. Acute attacks are associated commonly with use of certain medications, caloric deprivation, and changes in female reproductive hormones. About 20% of those with an acute attack also experience photosensitivity associated with bullae and skin fragility.
DIAGNOSIS/TESTING:
The most sensitive and specific biochemical screening test for any one of the acute porphyrias (including HCP) during an acute attack is a striking increase in urinary porphobilinogen (PBG). Quantitative analysis of porphyrins in both urine and feces is essential to distinguish between the different acute porphyrias and establish the diagnosis of HCP. Identification of a heterozygous pathogenic variant inCPOX (encoding the enzyme coproporphyrinogen-III oxidase) confirms the diagnosis and enables family studies.
MANAGEMENT:
Treatment of manifestations: Acute attacks are treated by discontinuation of any medications thought to induce attacks, management of dehydration and/or hyponatremia, administration of carbohydrate, and infusion of hematin. Treatment of symptoms and complications should be with medications known to be safe in acute porphyria (see www.drugs-porphyria.org). A minority of affected individuals experience repeat acute attacks, in which case management strategies include suppression of ovulation in females, prophylactic use of hematin, and liver transplantation when attacks and neurologic complications persist despite multiple courses of hematin. Prevention of primary manifestations: Agents or circumstances that may trigger an acute attack (including use of oral contraception in women) are avoided. Suppression of menses using a GnRH agonist (leuprolide, nafarelin, and others) may help CPOX heterozygotes who experience monthly exacerbations. Prevention of secondary complications: In CPOX heterozygotes undergoing surgery, intravenous glucose is provided in the perioperative period and non-barbiturate agents are used for induction of anesthesia. Agents/circumstances to avoid: Fasting, use of female reproductive hormones, and certain drugs including barbiturates and phenytoin. Evaluation of relatives at risk: If the family-specific CPOXpathogenic variant is known, clarification of the genetic status of relatives at risk allows early diagnosis of heterozygotes and education regarding how to avoid risk factors known to be associated with acute attacks.
GENETIC COUNSELING:
HCP is inherited in an autosomal dominant manner with reduced penetrance. Most individuals with HCP have an affected parent; the proportion with a de novo pathogenic variant is unknown. Each child of an individual with HCP has a 50% chance of inheriting the CPOX pathogenic variant. Because of reduced penetrance, many individuals with a CPOX pathogenic variant have no signs or symptoms of HCP. Prenatal diagnosis for pregnancies at increased risk is possible if the pathogenic variant in an affected family member is known.
Porphyria cutanea tarda (PCT) is a term encompassing a group of acquired and familial disorders in which activity of the heme synthetic enzyme uroporphyrinogen decarboxylase (UROD) is deficient.Approximately 80% of all cases of porphyria cutanea tarda are acquired; 20% are familial, although the ratio may vary among different geographic regions and ethnic groups.
Familial porphyria cutanea tarda most often arises from autosomal dominant inheritance of a single mutation of the UROD gene. Human UROD has been mapped to band 1p34.To date, 121 UROD mutations are listed by the Human Genome Mutation Database. A rare recessive familial type of porphyria cutanea tarda in which both UROD alleles are mutated is termed hepatoerythropoietic porphyria.Familial porphyria cutanea tarda without detectable UROD mutations has been reported.
The common acquired form, sporadic porphyria cutanea tarda, occurs in individuals whose UROD DNA sequences are normal, but who may have other genetically determined susceptibilities to inhibition of UROD activity. Acquired porphyria in large populations exposed to polyhalogenated aromatic hydrocarbon hepatotoxinshas been referred to as "epidemic porphyria cutanea tarda. Hepatic tumors producing excess porphyrins are rare causes of porphyria cutanea tardalike disorders.
Clinical expression of both sporadic and familial porphyria cutanea tarda most often requires exposure to environmental or infectious agents or the presence of coexisting conditions that adversely affect hepatocytes and result in hepatic siderosis. Ethanol intake, estrogen therapies, hemochromatosis genes, and hepatitis and human immunodeficiency viral infections are among these contributory factors.The increased oxidative stress associated with all of these factors has been shown to reduce hepatic expression of the gene encoding hepcidin, a regulator of iron absorption and metabolism, thus increasing iron absorption and iron overload.Excess iron facilitates formation of toxic oxygen species, thus amplifying porphyrinogenesis by catalyzing formation of oxidative inhibitors of UROD enzyme activity.Accumulating porphyrins in hepatocytes may then further down-regulate hepcidin gene expression. Most patients with porphyria cutanea tarda have increased iron burden; iron-reduction therapies can lead to clinical and biochemical remissions; subsequent reaccumulation of iron stores may lead to symptomatic recurrence.
Reduced UROD activity causes polycarboxylated porphyrinogen intermediaries of heme synthesis to accumulate in hepatocytes; these excess substrates then undergo iron-facilitated spontaneous oxidization to photoactive porphyrins. Porphyrin by-products of the pathway exit the hepatocytes, are distributed throughout the body in blood plasma, mediate photooxidative chemical reactions causing skin lesions, and yield the abnormal excretory porphyrin profiles that characterize porphyria cutanea tarda. Partial oxidation of uroporphyrinogen to the UROD inhibitor uroporphomethene occurs in murine porphyria cutanea tarda models and has been suggested as a pathogenic mechanism in the human disease.[9] Reduction of hepatic UROD activity to approximately 25% of normal, most often reflecting effects of multiple genetic and/or exogenous inhibitory factors, is required for clinical disease expression.
"Remember.Research is the key to your cure!"
Lina Rebeiz and her personal journey with AIP
Friday - September 16, 2016 @ 14:35:54
My porphyria symptoms started right after orientation of my freshman year of college. The pain started in my lower back and quickly moved to my abdomen, and within an hour I was curled up in my bed crying. My initial thought was that this must be food poisoning, and that my Resident Advisor's suggestion to go to the ER was ridiculous. When I woke up the next day, the pain had doubled and I began vomiting uncontrollably. I went directly to our student health and wellness center, where I was given Urinary Tract Infection (UTI) medication and advised to visit the ER if my symptoms worsened.
As the pain continued to intensify, I could not believe that this sensation was humanly possible. As soon as I thought the pain had reached its absolute limit, it would double. I finally decided to go to the ER, where I waited hours before being seen, only to be told that I should hydrate and relax for my UTI to go away. Nevertheless, because of the intensity of my pain, I was admitted to the hospital overnight while the doctors ran tests. When everything came back negative, I was sent home and told to go about my life normally. My parents, who had come to visit me while I was in the hospital, both scratched their heads at my weird symptoms. Though they are both physicians, they could not think of what could possibly be causing this misery.
After being discharged from the hospital un-diagnosed, I returned to the health and wellness center every day (sometimes multiple times a day) in the hopes that somebody would figure out what was wrong with me. I couldnt go to the hospital because they had already discharged me without a diagnosis. The pain was too unbearable to attend class, my parents made me feel guilty for staying home to rest, and everyone considered me an over-dramatic hypochondriac. Doctors repeated appendicitis and UTI tests, and every day they would tell me again that nothing was wrong with me. When I would call my parents crying, they would insist that I should trust the doctors and just relax. Unsure of what to do, I stopped going to class altogether because standing up and walking a few feet felt like running a marathon.
Over the course of a few weeks I lost more than 20 pounds; anything I tried to consume or drink (including water) would instantly be thrown up. After a month of being told that the symptoms were in my head, I started to believe the doctors. I stopped talking to my friends because I could not explain what was wrong with me; I stopped calling my parents and just retreated into my room until I eventually started getting better on my own.
Several months later, while on winter vacation with my family, my symptoms returned with a vengeance after I started taking birth control pills. My family, convinced that I had suffered something traumatic that I didnt want to share, insisted that I should take a break from school until I got better. I knew that this suggestion was ridiculous, and that there had to be something physically wrong with me. But without any medical evidence nobody believed me, and so we kept ignoring my symptoms.
After another two weeks, I again became malnourished -- but this time I also became delusional. My blood pressure had risen so high and my sodium had dropped so low that I suffered two seizures. During this episode, all I remember is the recurring feeling of insanity. I experienced mild hallucinations paired with strong delusions, and I always felt confused and scared. I had a constant feeling of worry, but not about anything concrete. Though it sound ridiculous now, I felt like something abstract, perhaps a monster, was out to get me.
It was during this episode that I was finally diagnosed. After the seizures, I was being treated at Tufts Medical Center, where my dad works; I am sure that without his stature as head of the otolaryngology department I would have been ignored once more. It took a whole team of doctors to figure out what was causing my symptoms; with the seizures and the PRES syndrome that followed, we finally had proof that these symptoms could not have been psychosomatic. With a stroke of luck, they sent out the test for Porphyria, and within a week the results came back positive.
When I received Panhematin, I bounced back within two days. After months of hell, I cannot put into words how relieved I was to receive an official diagnosis, and to start treatment. Now, having a diagnosis of Acute Intermittent Porphyria, I am confident that my symptoms will never be as extreme as they were then. I still do have sporadic attacks, but I am now able to recognize them and receive treatment early on. Though every day I wake up and wonder if that dull ache in my stomach, or that abstract feeling of anxiety means that a full-blown attack is coming, I have learned to manage the pain and to take it one day at a time.
Learning about Porphyria
Friday - September 9, 2016 @ 10:00:13
Learning about Porphyria
To view entire link: https://www.genome.gov/19016728/learning-about-porphyria/
The porphyrias are a group of different diseases, each caused by a specific abnormality in the heme production process. Heme is a chemical compound that contains iron and gives blood its red color. The essential functions of heme depend on its ability to bind oxygen. Heme is incorporated into hemoglobin, a protein that enables red blood cells to carry oxygen from the lungs to all parts of the body. Heme also plays a role in the liver where it assists in breaking down chemicals (including some drugs and hormones) so that they are easily removed from the body.
Heme is produced in the bone marrow and liver through a complex process controlled by eight different enzymes. As this production process of heme progresses, several different intermediate compounds (heme precursors) are created and modified. If one of the essential enzymes in heme production is deficient, certain precursors may accumulate in tissues (especially in the bone marrow or liver), appear in excess in the blood, and get excreted in the urine or stool. The specific precursors that accumulate depend on which enzyme is deficient. Porphyria results in a deficiency or inactivity of a specific enzyme in the heme production process, with resulting accumulation of heme precursors.
What are the signs and symptoms of porphyria?
The signs and symptoms of porphyria vary among types. Some types of porphyria (called cutaneous porphyria) cause the skin to become overly sensitive to sunlight. Areas of the skin exposed to the sun develop redness, blistering and often scarring.
The symptoms of other types of porphyria (called acute porphyrias) affect the nervous system. These symptoms include chest and abdominal pain, emotional and mental disorders, seizures and muscle weakness. These symptoms often appear quickly and last from days to weeks. Some porphyrias have a combination of acute symptoms and symptoms that affect the skin.
Environmental factors can trigger the signs and symptoms of porphyria. These include:
Alcohol
Smoking
Certain drugs, hormones
Exposure to sunlight
Stress
Dieting and fasting
How is porphyria diagnosed?
Porphyria is diagnosed through blood, urine, and stool tests, especially at or near the time of symptoms. Diagnosis may be difficult because the range of symptoms is common to many disorders and interpretation of the tests may be complex. A large number of tests are available, however, but results among laboratories are not always reliable.
How is porphyria treated?
Each form of porphyria is treated differently. Treatment may involve treating with heme, giving medicines to relieve the symptoms, or drawing blood. People who have severe attacks may need to be hospitalized.
What do we know about porphyria and heredity?
Most of the porphyrias are inherited conditions. The genes for all the enzymes in the heme pathway have been identified. Some forms of porphyria result from inheriting one altered gene from one parent (autosomal dominant). Other forms result from inheriting two altered genes, one from each parent (autosomal recessive). Each type of porphyria carries a different risk that individuals in an affected family will have the disease or transmit it to their children.
Porphyria cutanea tarda (PCT) is a type of porphyria that is most often not inherited. Eighty percent of individuals with PCT have an acquired disease that becomes active when factors such as iron, alcohol, hepatitis C virus (HCV), HIV, estrogens (such as those used in oral contraceptives and prostate cancer treatment), and possibly smoking, combine to cause an enzyme deficiency in the liver. Hemochromatosis, an iron overload disorder, can also predispose individuals to PCT. Twenty percent of individuals with PCT have an inherited form of the disease. Many individuals with the inherited form of PCT never develop symptoms.
If you or someone you know has porphyria, we recommend that you contact a genetics clinic to discuss this information with a genetics professional. To find a genetics clinic near you, contact your primary doctor for a referral.
What triggers a porphyria attack?
Porphyria can be triggered by drugs (barbiturates, tranquilizers, birth control pills, sedatives), chemicals, fasting, smoking, drinking alcohol, infections, emotional and physical stress, menstrual hormones, and exposure to the sun. Attacks of porphyria can develop over hours or days and last for days or weeks.
How is porphyria classified?
The porphyrias have several different classification systems. The most accurate classification is by the specific enzyme deficiency. Another classification system distinguishes porphyrias that cause neurologic symptoms (acute porphyrias) from those that cause photosensitivity (cutaneous porphyrias). A third classification system is based on whether the excess precursors originate primarily in the liver (hepatic porphyrias) or primarily in the bone marrow (erythropoietic porphyrias). Some porphyrias are classified as more than one of these categories.
What are the cutaneous porphyrias?
The cutaneous porphyrias affect the skin. People with cutaneous porphyria develop blisters, itching, and swelling of their skin when it is exposed to sunlight. The cutaneous porphyrias include the following types:
Also called congenital porphyria. This is a rare disorder that mainly affects the skin. It results from low levels of the enzyme responsible for the fourth step in heme production. It is inherited in an autosomal recessive pattern.
An uncommon disorder that mainly affects the skin. It results from reduced levels of the enzyme responsible for the eighth and final step in heme production. The inheritance of this condition is not fully understood. Most cases are probably inherited in an autosomal dominant pattern, however, it shows autosomal recessive inheritance in a small number of families.
A rare disorder that mainly affects the skin. It results from very low levels of the enzyme responsible for the fifth step in heme production. It is inherited in an autosomal recessive pattern.
A rare disorder that can have symptoms of acute porphyria and symptoms that affect the skin. It results from low levels of the enzyme responsible for the sixth step in heme production. It is inherited in an autosomal dominant pattern.
The most common type of porphyria. It occurs in an estimated 1 in 25,000 people, including both inherited and sporadic (noninherited) cases. An estimated 80 percent of porphyria cutanea tarda cases are sporadic. It results from low levels of the enzyme responsible for the fifth step in heme production. When this condition is inherited, it occurs in an autosomal dominant pattern.
A disorder that can have symptoms of acute porphyria and symptoms that affect the skin. It results from low levels of the enzyme responsible for the seventh step in heme production. It is inherited in an autosomal dominant pattern.
The acute porphyrias affect the nervous system. Symptoms of acute porphyria include pain in the chest, abdomen, limbs, or back; muscle numbness, tingling, paralysis, or cramping; vomiting; constipation; and personality changes or mental disorders. These symptoms appear intermittently. The acute porphyrias include the following types:
This is probably the most common porphyria with acute (severe but usually not long-lasting) symptoms. It results from low levels of the enzyme responsible for the third step in heme production. It is inherited in an autosomal dominant pattern.
A very rare disorder that results from low levels of the enzyme responsible for the second step in heme production. It is inherited in an autosomal recessive pattern.
It is time to reserve your hotel room for the FDA meeting! The APF has secured a room block at the special rate of $99/night at the Holiday Inn College Park (10000 Baltimore Avenue, College Park, MD 20740). The room block discount is available for Sunday, October 23rd and Monday, October 24th. The hotel currently has rooms available surrounding these dates, should you need them, but they are not at this discounted rate.
To book your room, please call the hotel directly at 301-345-6700 and ask to be transferred to Central Reservations. You may also contact the hotel via email: reservations@hicollegepark.com. In order to receive the special rate, you MUST say you are with the AMERICAN PORPHYRIA FOUNDATION at the time of booking.
The official deadline to reserve is 9/23/2016, but make sure to book your room ASAP. Rooms are first come, first serve. If the room block fills up, we will request additional rooms, but they are not guaranteed. You are absolutely not required to stay at this hotel, but this is the location of the APF meeting on Sunday, October 23rd. If you have not already, please make sure you RSVP to me for both the APF meeting and FDA meeting.
Please contact me if you have any questions! We are looking forward to meeting everyone!
Chicago Patient Education Meeting
Thursday - September 1, 2016 @ 15:04:32
The invitations have gone out for the Chicago Patient Education Meeting on September 24th! APF member Ruth Dee Bruno will be hosting this meeting and we hope you will take advantage of this opportunity to meet new friends who share your experiences with porphyria. There will be presentations and a Q & A session for all attendees. Bring your friends and family! We can't wait to see you all there! Saturday, September 24, 2016 5:30PM - 7:30PM CDT 2509 Mill Creek Lane Rolling Meadows, Illinois 60008
Seating is limited - Please RSVP by email: colorimage1@yahoo.com or by phone: 630-450-6769
**If you would like to host a patient meeting of your own, let the APF
know! We will assist you in planning and advertising for your meeting, as well as supply APF materials.**
Happy First Day Of September
Thursday - September 1, 2016 @ 14:57:04
Are you ready for Fall? Winter? Curl up with a good book, stay healthy and well! The American Porphyria Foundation thanks all of those who are willing to attend the FDA, getting involved in research opportunities. Your support and love for one another on the sites. Thank you for all the wonderful videos, donations, and hosting the APF meetings, the runners, the wristband and tshirt supports. You all make a difference. Keep fighting so that one day we may all have a cure!
Key Terms- How do they relate to porphyria? Check the article at this link: http://www.encyclopedia.com/topic/Porphyrias.aspx
Monday - August 29, 2016 @ 10:30:07
Key Terms- How do they relate to porphyria? Check the article at this link: http://www.encyclopedia.com/topic/Porphyrias.aspx
Terms:
Autosomal dominant A pattern of inheritance in which only one of the two copies of an autosomal gene must be abnormal for a genetic condition or disease to occur. An autosomal gene is a gene that is located on one of the autosomes or non-sex chromosomes. A person with an autosomal dominant disorder has a 50 percent chance of passing it to each of their offspring.
Autosomal recessive A pattern of inheritance in which both copies of an autosomal gene must be abnormal for a genetic condition or disease to occur. An autosomal gene is a gene that is located on one of the autosomes or non-sex chromosomes. When both parents have one abnormal copy of the same gene, they have a 25 percent chance with each pregnancy that their offspring will have the disorder.
Biosynthesis The manufacture of materials in a biological system.
Bone marrow The spongy tissue inside the large bones in the body that is responsible for making the red blood cells, most white blood cells, and platelets.
Chromosome A microscopic thread-like structure found within each cell of the human body and consisting of a complex of proteins and DNA. Humans have 46 chromosomes arranged into 23 pairs. Chromosomes contain the genetic information necessary to direct the development and functioning of all cells and systems in the body. They pass on hereditary traits from parents to child (like eye color) and determine whether the child will be male or female.
Enzyme A protein that catalyzes a biochemical reaction without changing its own structure or function.
Erythropoiesis The process through which new red blood cells are created; it begins in the bone marrow.
Erythropoietic Referring to the creation of new red blood cells.
Gene A building block of inheritance, which contains the instructions for the production of a particular protein, and is made up of a molecular sequence found on a section of DNA. Each gene is found on a precise location on a chromosome.
Hematin A drug administered intravenously to halt an acute porphyria attack. It causes heme biosynthesis to decrease, preventing the further accumulation of heme precursors.
Heme The iron-containing molecule in hemoglobin that serves as the site for oxygen binding.
Hemoglobin An iron-containing pigment of red blood cells composed of four amino acid chains (alpha, beta, gamma, delta) that delivers oxygen from the lungs to the cells of the body and carries carbon dioxide from the cells to the lungs.
Hepatic Refers to the liver.
Neuropathy A disease or abnormality of the peripheral nerves (the nerves outside the brain and spinal cord). Major symptoms include weakness, numbness, paralysis, or pain in the affected area.
Porphyrin An organic compound found in living things that founds the foundation structure for hemoglobin, chlorophyll, and other respiratory pigments. In humans, porphyrins combine with iron to form hemes.
Protoporphyrin A kind of porphyrin that links with iron to form the heme of hemoglobin
"Remember.Research is the key to your cure!"
Rare Disease United Foundations Beyond the Diagnosis Art Exhibit
Tuesday - August 23, 2016 @ 10:30:32
We would like to share that Rare Disease United Foundation's Beyond the Diagnosis Art Exhibit will be the headline story on CBS News Sunday Morning with Charles Osgood airing this Sunday, August 28th at 9:00am EST. We are very excited they are bringing national attention to rare diseases and the issues surrounding rare diseases. This group now has master artists from around the world donating their time and talent to paint children with rare diseases. Their goal is to put a face to all 7,000 known rare diseases.
The Rare Disease United Foundation is a non-disease speci�c, community-based organization, working at a state-level on legislation that has a direct impact on people living with a rare disease, providing support locally, and establishing relationships at local hospitals and medical schools.
For more information about the Rare Disease United Foundation, please visit http://rarediseaseunited.org/ or email at info@rarediseaseunited.org.
RSVP FOR EPP MEETING with the FDA 10/24/16 Info
Friday - August 19, 2016 @ 10:30:05
Hello everyone! We are very excited about the number of people we have already had RSVP for the upcoming meeting at the FDA. While we finalize the details of the meeting, here are a few things to keep in mind. PLEASE read everything carefully and feel free to contact me if you have ANY questions!
THINGS TO KNOW FOR THE FDA MEETING FOR EPP:
1. The FDA Meeting is Monday, October 24th from 10AM 4PM. The meeting will last the entire time, please plan your travel accordingly.
2. RSVPing to the APF DOES NOT register you for the FDA meeting. MAKE SURE you are registering at THIS LINK by October 17th, 2016. The same link is used for registering in person and via webcast.
3. Make sure your valid Drivers License, State ID or USA Passport matches the name you register with! You will need to show proof of ID before entering the FDA meeting. If you have already registered and it does not, or you are unsure, contact the event organizer at meghana.chalasani@fda.hhs.gov.
4. If you have signed up for the meeting with the FDA, you should have received a confirmation email from EventBrite summarizing your registration information.
5. If you have ALSO signed up to participate in the panel during the meeting, you should have received a SECOND email from Meghana Chalasani (FDA meeting coordinator) with a list of discussion questions. The deadline to submit your reply for the panel is October 10th, 2016. You will be notified of your panel status at least a week in advance of the meeting. If you have signed up to speak at the panel, but did not receive this email, then you are not being considered for the panel. You must contact Ms. Chalasani to receive the questions.
6. The APF will be securing a room block for Sunday, October 23rd and Monday, October 24th at the Holiday Inn Washington College Park. This is also where the APF meeting will be on Sunday, October 23rd.
Address: 10000 Baltimore Ave, College Park, MD 20740
The room block is NOT finalized at this time. We need to have an idea of how many of you will be using a room in the block and for which nights. Please let me know ASAP so we can sign the contract and finalize the hotel.
7. The APF meeting, with Dr. Robert Desnick, will be Sunday, October 23rd from 5PM 7PM at the Holiday Inn College Park. The room will be available starting at 2PM as an informal meeting space for attendees to mingle and get to know each other! Stop by and say hi to your fellow EPPers!
If you have ANY questions, or just want to let me know how excited you are to attend, dont hesitate to contact me! My email is Jessica@porphyriafoundation.org. We are so excited this is happening and I look forward to meeting you all in person (finally)! :)
HEP Type of Porphyria Read & Share
Monday - August 15, 2016 @ 10:30:14
Hepatoerythropoietic Porphyria (HEP)
What is Hepatoerythropoietic Porphyria?
HEP is a deficiency of the enzyme uroporphyrinogen decarboxylase; it is the autosomal recessive form of f-PCT. The manifestations of HEP resemble Congenital Erythropoietic Porphyria (CEP), with symptoms of skin blistering that usually begin in infancy.
Skin photosensitivity results in severe blistering and scarring, often with mutilation and loss of facial features and fingers. Increased hair growth (hypertrichosis) on sun-exposed skin, brownish-colored teeth (erythrodontia), and reddish-colored urine are common. There may be bone fragility due to expansion of the bone marrow and vitamin deficiencies, especially vitamin D. Red blood cells have a shortened life-span, and mild or severe hemolytic anemia often results. Synthesis of heme and hemoglobin is actually increased to compensate for the shortened red blood cell survival and is associated with splenomegaly. Bacteria may infect the damaged skin and contribute to mutilation and scarring.
Who gets Hepatoerythropoietic Porphyria?
HEP is a very rare type of autosomal recessive porphyria. Each parent of an affected individual must have a mutation in one of their UROD genes and both must pass their mutation on to their child. This also means that both parents have f-PCT.
What causes Hepatoerythropoietic Porphyria?
HEP is caused by a deficiency of the enzyme uroporphyrinogen decarboxylase, due to the inheritance of mutations in both copies of a persons URO-decarboxylase genes.
How is Hepatoerythropoietic Porphyria diagnosed?
Diagnosis of HEP can be made by demonstrating significant elevations of specific porphyrins in urine and stool, as well as iidentification of a specific fluorescence emission peak in plasma. DNA testing to identify the specific mutations in an individuals UROD genes is the most specific and sensitive test to confirm the diagnosis of HEP.
What are treatments for Hepatoerythropoietic Porphyria?
Treatment is the same as for PCT: regularly scheduled phlebotomies (removal of blood) to lower the amount of porphyrins in the liver or a low dose regimen of hydroxychloroquine as well as removal of factors (for example, certain medications) that activated the disease and avoidance and/or protection from sunlight.
Additional Reading about HEP:
Hepatoerythropoietic Porphyria
NORD gratefully acknowledges Ashwani K Singal, MD, MSc, Division of Gastroenterology and Hepatology, University of Alabama at Birmingham, for assistance in the preparation of this report.
Synonyms of Hepatoerythropoietic Porphyria
autosomal recessive PCT
HEP
General Discussion
Summary
Hepatoerythropoietic porphyria (HEP) is an extremely rare genetic disorder characterized by deficiency of the enzyme, uroporphyrinogen decarboxylase. This deficiency is caused by mutations of both copies of a persons UROD gene, which means that the disorder is inherited as an autosomal recessive trait. Most affected individuals have a profound deficiency of this enzyme and onset of the disorder is usually during infancy or early childhood. However, some individuals may have a mild form that can go undiagnosed until adulthood. The childhood form of HEP is often associated with painful, blistering skin lesions that develop on sun-exposed skin (photosensitivity). Affected areas of skin can scar and become discolored. There may be risk of bacterial infection. Abnormal, excessive hair (hypertrichosis) on affected skin is also common. Mild anemia and abnormal enlargement of the liver and/or spleen (hepatosplenomegaly) have also been reported. Mild cases of HEP may go unrecognized until adulthood and can be clinically indistinguishable from porphyria cutanea tarda (PCT), a related disorder that may be acquired or occur in individuals with a mutation of one UROD gene (autosomal dominant inheritance). Cutaneous photosensitivity is generally much more severe in HEP than in PCT. NORD has a separate report on porphyria cutanea tarda.
Introduction
HEP belongs to a group of disorders known as the porphyrias. This group of at least seven disorders is characterized by abnormally high levels of porphyrins and porphyrin precursors due to deficiency of certain enzymes essential to the creation (synthesis) of heme, a part of hemoglobin and other hemoproteins. There are eight enzymes in the pathway for making heme and at least seven major forms of porphyria. The symptoms associated with the various forms of porphyria differ. It is important to note that people who have one type of porphyria do not develop any of the other types. Porphyrias are generally classified into two groups: the "hepatic" and "erythropoietic" types. Porphyrins and porphyrin precursors and related substances originate in excess amounts predominantly from the liver in the hepatic types and mostly from the bone marrow in the erythropoietic types. Porphyrias with skin manifestations are sometimes referred to as "cutaneous porphyrias". The term "acute porphyria" is used to describe porphyrias that can be associated with sudden attacks of pain and other neurological symptoms. HEP is a hepatic and cutaneous porphyria.
Signs & Symptoms
The symptoms and severity of HEP can vary from one person to another. Onset is usually within the first two years of life, but mild cases that go undiagnosed until adulthood have been reported. Although HEP is associated with specific, characteristic symptoms, several factors, including the small number of identified cases, make it difficult to establish the full range of associated symptoms of the disorder.
Severe cutaneous photosensitivity is usually the first sign. Affected infants may have extremely fragile skin that that can peel or blister on minimal impact is common. Reddening of the skin is common (erythema). Blistering skin lesions can develop on sun-exposed skin such as the hands and face. Photosensitivity can be severe and can cause scarring, erosion, and disfigurement. Bacterial infection of skin lesions can occur.
Abnormal, excessive hair growth (hypertrichosis) may also occur on sun-exposed skin. Affected skin may darken or lose color (hyper- or hypopigmentation). Small bumps with a distinct white head (milia) may also develop. Some affected individuals have teeth that are reddish-brown colored (erythrodontia).
Low levels of circulating red blood cells (anemia) may also occur. Anemia may be due to the premature destruction of red blood cells (hemolysis). Anemia associated with HEP may be mild or severe. Severe anemia may be associated with fatigue, pale skin, irregular heartbeat, chest pain, dizziness, and abnormally cold hands and feet. Some individuals may have an abnormally enlarged liver and/or spleen (hepatosplenomegaly).
Mild cases of HEP can go undiagnosed until adulthood. Overt photosensitivity may not be seen and mild skin damage can be mistaken for other conditions during childhood.
Causes
HEP is caused by mutations of both alleles of the UROD gene. Genes provide instructions for creating proteins that play a critical role in many functions of the body. When a mutation of a gene occurs, the protein product may be faulty, inefficient, or absent. Depending upon the functions of the particular protein, this can affect many organ systems of the body.
HEP is inherited as an autosomal recessive trait. Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Recessive genetic disorders occur when an individual inherits the same abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
Investigators have determined that the UROD gene is located on the short arm (p) of chromosome 1 (1p34.1). Chromosomes, which are present in the nucleus of human cells, carry the genetic information for each individual. Human body cells normally have 46 chromosomes. Pairs of human chromosomes are numbered from 1 through 22 and the sex chromosomes are designated X and Y. Males have one X and one Y chromosome and females have two X chromosomes. Each chromosome has a short arm designated p and a long arm designated q. Chromosomes are further sub-divided into many bands that are numbered. For example, chromosome 1p34.1 refers to band 34.1 on the short arm of chromosome 1. The numbered bands specify the location of the thousands of genes that are present on each chromosome.
The UROD gene creates (encodes) an enzyme known as uroporphyrinogen decarboxylase (UROD), which is the fifth enzyme in the heme biosynthetic pathway. In HEP, UROD enzyme activity is usually less than 10% its normal levels. Such low enzyme activity results in the abnormal accumulation of specific porphyrins and related chemicals in body, especially within the bone marrow, red blood cells, liver and skin. Symptoms develop because of this abnormal accumulation of porphyrins and related chemicals. For example when porphyrins accumulate in the skin, they absorb sunlight and enter an excited state (photoactivation). This abnormal activation results in the characteristic damage to the skin found in individuals with HEP. The liver removes porphyrins from the blood plasma and secretes it into the bile. When porphyrins accumulate in the liver, they can cause toxic damage to the liver.
Affected Populations
HEP is an extremely rare disorder that affects males and females in equal numbers. Approximately 40 cases have been reported in the medical literature. The exact incidence or prevalence of HEP in the general population is unknown.
Related Disorders
Symptoms of the following disorders can be similar to those of HEP. Comparisons may be useful for a differential diagnosis.
Congenital erythropoietic porphyria (CEP) is a rare inherited metabolic disorder resulting from the deficient function of the enzyme uroporphyrinogen III cosynthase (UROS), the fourth enzyme in the heme biosynthetic pathway. Due to the impaired function of this enzyme, excessive amounts of particular porphyrins accumulate, particularly in the bone marrow, plasma, red blood cells, urine, teeth, and bones. The major symptom of this disorder is hypersensitivity of the skin to sunlight and some types of artificial light, such as fluorescent lights (photosensitivity). After exposure to light, the photo-activated porphyrins in the skin cause bullae (blistering) and the fluid-filled sacs rupture, and the lesions often get infected. These infected lesions can lead to scarring, bone loss, and deformities. The hands, arms, and face are the most commonly affected areas. CEP is inherited as an autosomal recessive genetic disorder. Typically, there is no family history of the disease. Both parents are usually healthy, but each carries a defective gene that they can pass to their children. Affected offspring have two copies of the defective gene, one inherited from each parent. (For more information on this disorder, choose congenital erythropoietic porphyria as your search term in the Rare Disease Database.)
There are other conditions that may cause signs and symptoms that are similar to those seen in HEP. Such conditions include other cutaneous porphyrias, drug-induced photosensitivity, epidermolysis bullosa, various forms of lupus, and solar urticarial. (For more information on these disorders, choose the specific disorder name as your search term in the Rare Disease Database.)
Diagnosis
A diagnosis of HEP is based upon identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation and a variety of specialized tests. HEP may be considered in infants and children with chronic, blistering photosensitivity.
Clinical Testing and Workup
Screening tests can help diagnose HEP by measuring the levels of certain porphyrins in blood plasma, urine and red blood cells. These tests can help to differentiate the disorder from congenital erythropoietic porphyria by the different patterns of individual porphyrins and/or by demonstrating markedly decreased activity of the UROD enzyme. There is elevation of porphyrins in plasma, urine, and feces. Porphyrin patterns in HEP are similar to those seen in PCT with elevation of highly carboxylated porphyrins and isocoproporphyrins. In contrast to PCT, there are markedly increased levels of zinc protoporphyrin in red blood cells in HEP patients which is due to accumulation of pathway intermediates being metabolized to protoporphyrins.
Molecular genetic testing can confirm a diagnosis of HEP by detecting mutations in both UROD genes, but is available only on a clinical basis.
Standard Therapies
Treatment
The treatment of HEP is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, hematologists, dermatologists, hepatologists, and other healthcare professionals may need to systematically and comprehensively plan an affected childs treatment. Genetic counseling may benefit affected individuals and their families.
There is no specific, FDA-approved therapy for individuals with HEP. Because the disorder is so rare, most treatment information is based other forms of porphyria.
Avoidance of sunlight will benefit affected individuals and can include the use of clothing styles with long sleeves and pant legs, made with double layers of fabric or of light-exclusive fabrics, wide brimmed hats, gloves, and sunglasses. Topical sunscreens are generally ineffective, but certain tanning products with ingredients that increase pigmentation may be helpful. Affected individuals may also benefit from window tinting and the use of vinyl or films to cover the windows of their homes and cars.
Phlebotomies, which are used to treat individuals with PCT, are generally ineffective in individuals with HEP since elevated iron levels are not a feature of the disorder. Another treatment for PCT, the antimalarial drug chloroquine, was effective in at least one case reported in the medical literature.
Anemia may require treatment in some cases. Blood transfusions have been used to treat some individuals. Recombinant erythropoietin, which helps the body produce more red blood cells, was successfully used to treat severe anemia in an individual with HEP whose anemia was not associated with increased red cell destruction.
Investigational Therapies
Gene therapy is also being studied as another approach to therapy for individuals with genetic disorder associated with enzyme deficiency. In gene therapy, the defective gene present in a patient is replaced with a normal gene to enable the produce of the active enzyme and prevent the development and progression of the disease in question. Given the permanent transfer of the normal gene, which is able to produce active enzyme at all sites of disease, this form of therapy is theoretically most likely to lead to a cure. However, at this time, there remain some technical difficulties to resolve before gene therapy can be advocated as a viable alternative approach for genetic disorders like HEP.
To-Figueras J, Phillips JD, Gonzalez-Lopez JM, et al. Hepatoeythropoetic porphyria due to a novel mutation in the uroporphyrinogen decarboxylase gene. Br J Dermatol. 2011;165:499-505.http://www.ncbi.nlm.nih.gov/pubmed/21668429
Cantatore-Francis JL, Cohen J, Balwani M, et al. Hepatoerythropoietic porphyria misdiagnosed as child abuse: cutaneous, arthritic, and hematologic manifestations in siblings with a novel UROD mutation. Arch Dermatol. 2010;146:529-533. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3092549/
Armstrong DK, Sharpe PC, Chambers CR, et al. Hepatoerythropoietic porphyria: a missense mutation in the UROD gene is associated with mild disease and an unusual porphyrin excretion pattern. Br J Dermatol. 2004;151:920-923. http://www.ncbi.nlm.nih.gov/pubmed/15491440
Ged C, Ozalla D, Herrero C, et al. Description of a new mutation in hepatoerythropoietic porphyria and prenatal exclusion of a homozygous fetus. Arch Dermatol. 2002;138:957-960.http://www.ncbi.nlm.nih.gov/pubmed/12071824
Moran-Jimenez MJ, Ged C, Romana M, et al. Uroporphyrinogen decarboxylase: complete human gene sequence and molecular study of three families with hepatoerythropoietic porphyria. Am J Hum Genet. 1996;58:712-721. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1914669/
The APF would like to extend wholehearted congratulations to esteemed porphyria expert, Dr. Peter Tishler, on his recent retirement. Dr. Tishler has been a treasured member of the APF and our Scientific Advisory Board for many years and we THANK YOU for your invaluable service to the porphyria community!
We welcome his colleague, Dr. Joel Krier, to the APF. Dr. Krier has been mentored by Dr. Tishler and we look forward to working with him. If you are looking for a porphyria expert in the Massachusetts area, please contact the APF and we will gladly put you in touch with Dr. Krier.
Thank you, Dr. Tishler! We wish you the best!
"Remember.Research is the key to your cure!"
APF Announcements
Friday - August 12, 2016 @ 10:30:26
The American Porphyria Foundation wants YOU to be aware that there are many trials going on in the Porphyria community. This is critical to learning more about the Porphyrias, and ultimately a cure. Current steps for instance for those who suffer from EPP have a chance to meet with the FDA for approval soon of a new drug. In addition, the Acute Porphyrias are being treatment for a new type of drug. We are always looking for those who are interested in participating in research answering questions, to travel and meeting with an expert or more importantly getting a better manageable system in place to manage your type of Porphyria. Please use this link to learn more about the trials at the below links: https://clinicaltrials.gov/ct2/results?cond=%22porphyria%22 http://rarediseases.org/ Also note: If you have not become a member of the American Porphyria Foundation why not do so today. Membership is free. If you would like to make an annual contribution to the APF of $30.00 you will benefit from the events, newsletters, enews and special announcements, meetings and patient gatherings. The APF is a non-profit organization so your participation and support will go a long way in continuing the many fine education, and PTF (protect the future doctors) Also we have many exciting items for sale you can look to the APF: www.porphyriafoundation.org We have wonderful EPP & Acute ED Emergency guidelines books also available for a small fee please contact the APF 1/866/apf/3635
If you have not received your free patient kit or asked for your physician comprehensive Dr. Kit we would be most happy to send those out to you and your Doctor. Please contact the APF 1/866/apf/3635 "Remember.Research is the key to your cure!"
Diagnosis & Management of the Porphyrias:Diagnosis & Management of the Porphyrias: Part 4
Tuesday - August 9, 2016 @ 10:30:40
Diagnosis & Management of the Porphyrias: Click on this link and tab to learn more about each type: Diagnosis & Management of the Porphyrias:
These resources address the diagnosis or management of porphyria:
Some types of porphyria are inherited in an autosomal dominant pattern, which means one copy of the gene in each cell is mutated. This single mutation is sufficient to reduce the activity of an enzyme needed for heme production, which increases the risk of developing signs and symptoms of porphyria. Autosomal dominant porphyrias include acute intermittent porphyria, most cases of erythropoietic protoporphyria, hereditary coproporphyria, and variegate porphyria. Although the gene mutations associated with some cases of porphyria cutanea tarda also have an autosomal dominant inheritance pattern, most people with this form of porphyria do not have an inherited gene mutation.
Other porphyrias are inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but do not show signs and symptoms of the condition. Porphyrias with an autosomal recessive pattern of inheritance include ALAD deficiency porphyria, congenital erythropoietic porphyria, and some cases of erythropoietic protoporphyria.
When erythropoietic protoporphyria is caused by mutations in the ALAS2 gene, it has an X-linked dominant pattern of inheritance. The ALAS2 gene is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the gene in each cell may be sufficient to cause the disorder. In males (who have only one X chromosome), a mutation in the only copy of the gene in each cell causes the disorder. Males may experience more severe symptoms of the disorder than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Mutations in the UROD gene are related to both porphyria cutanea tarda and hepatoerythropoieticporphyria. Individuals who inherit one altered copy of the UROD gene are at increased risk forporphyria cutanea tarda. (Multiple genetic and nongenetic factors contribute to this condition.) People who inherit two altered copies of the UROD gene in each cell develop hepatoerythropoieticporphyria.
Porphyria Genetic Part 2 https://ghr.nlm.nih.gov/condition/porphyria#genes Read & Share
Monday - August 1, 2016 @ 10:30:15
Frequency:
The exact prevalence of porphyria is unknown, but it probably ranges from 1 in 500 to 1 in 50,000 people worldwide. Overall, porphyria cutanea tarda is the most common type of porphyria. For some forms of porphyria, the prevalence is unknown because many people with a genetic mutation associated with the disease never experience signs or symptoms.
Acute intermittent porphyria is the most common form of acute porphyria in most countries. It may occur more frequently in northern European countries, such as Sweden, and in the United Kingdom. Another form of the disorder, hereditary coproporphyria, has been reported mostly in Europe and North America. Variegate porphyria is most common in the Afrikaner population of South Africa; about 3 in 1,000 people in this population have the genetic change that causes this form of the disorder.
The genes related to porphyria provide instructions for making the enzymes needed to produceheme. Mutations in most of these genes reduce enzyme activity, which limits the amount of heme the body can produce. As a result, compounds called porphyrins and porphyrin precursors, which are formed during the process of heme production, can build up abnormally in the liver and other organs. When these substances accumulate in the skin and interact with sunlight, they cause the cutaneous forms of porphyria. The acute forms of the disease occur when porphyrins and porphyrin precursors build up in and damage the nervous system.
One type of porphyria, porphyria cutanea tarda, results from both genetic and nongenetic factors. About 20 percent of cases are related to mutations in the UROD gene. The remaining cases are not associated with UROD gene mutations and are classified as sporadic. Many factors contribute to the development of porphyria cutanea tarda. These include an increased amount of iron in the liver, alcohol consumption, smoking, hepatitis C or HIV infection, or certain hormones. Mutations in theHFE gene (which cause an iron overload disorder called hemochromatosis) are also associated with porphyria cutanea tarda. Other, as-yet-unidentified genetic factors may also play a role in this form of porphyria.
Porphyria is a group of disorders caused by abnormalities in the chemical steps that lead to heme production. Heme is a vital molecule for all of the body's organs, although it is most abundant in the blood, bone marrow, and liver. Heme is a component of several iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood).
Researchers have identified several types of porphyria, which are distinguished by their genetic cause and their signs and symptoms. Some types of porphyria, called cutaneous porphyrias, primarily affect the skin. Areas of skin exposed to the sun become fragile and blistered, which can lead to infection, scarring, changes in skin coloring (pigmentation), and increased hair growth. Cutaneous porphyrias include congenital erythropoietic porphyria, erythropoietic protoporphyria, hepatoerythropoietic porphyria, and porphyria cutanea tarda.
Other types of porphyria, called acute porphyrias, primarily affect the nervous system. These disorders are described as "acute" because their signs and symptoms appear quickly and usually last a short time. Episodes of acute porphyria can cause abdominal pain, vomiting, constipation, and diarrhea. During an episode, a person may also experience muscle weakness, seizures, fever, and mental changes such as anxiety and hallucinations. These signs and symptoms can be life-threatening, especially if the muscles that control breathing become paralyzed. Acute porphyrias include acute intermittent porphyria and ALAD deficiency porphyria. Two other forms of porphyria, hereditary coproporphyria and variegate porphyria, can have both acute and cutaneous symptoms.
The porphyrias can also be split into erythropoietic and hepatic types, depending on where damaging compounds called porphyrins and porphyrin precursors first build up in the body. In erythropoietic porphyrias, these compounds originate in the bone marrow. Erythropoietic porphyrias include erythropoietic protoporphyria and congenital erythropoietic porphyria. Health problems associated with erythropoietic porphyrias include a low number of red blood cells (anemia) and enlargement of the spleen (splenomegaly). The other types of porphyrias are considered hepatic porphyrias. In these disorders, porphyrins and porphyrin precursors originate primarily in the liver, leading to abnormal liver function and an increased risk of developing liver cancer.
Environmental factors can strongly influence the occurrence and severity of signs and symptoms of porphyria. Alcohol, smoking, certain drugs, hormones, other illnesses, stress, and dieting or periods without food (fasting) can all trigger the signs and symptoms of some forms of the disorder. Additionally, exposure to sunlight worsens the skin damage in people with cutaneous porphyrias.
"Remember.Research is the key to your cure!"
The Dateline NBC documentary "Out of the Shadows" featuring EPP has been nominated for an Emmy Award!
Friday - July 22, 2016 @ 10:00:25
The Dateline NBC documentary "Out of the Shadows" featuring EPP has been nominated for an Emmy Award! The APF received this great message from an NBC producer:
I'm excited to report that the show that you helped build, Dateline NBC's "Out of the Shadows," was nominated for an Emmy Award for Best Feature in a News Magazine. I can't thank you and the entire EPP community enough for all your help. The ceremony is in September. Fingers crossed!
*The APF would like to thank everyone involved in the creation of this episode.
Visit this website to view the official announcement:
Don't forget to mark your calendars for the next APF Patient Meeting Indianapolis
Wednesday - July 20, 2016 @ 10:00:26
Next APF Patient Meeting Scheduled in Indianapolis
APF member Nichol Kirby invites you to a Patient Education Meeting in Indianapolis, IN! We hope you will take advantage of this opportunity to meet friends who share your experiences with porphyria. There will be a presentation and a Q & A session for all attendees.
Bring your friends and family! We can't wait to see you all there!
Saturday, July 30, 2016 11AM - 1PM EDT IU Health Simon Cancer Center Healing Garden 1030 W. Michigan St. Indianapolis, IN 46202
**Seating is limited so we ask that you please RSVP to Nichol at 317-749-7477 ornichol.kirby@gmail.com**
"Remember.Research is the key to your cure!"
FDA accepts SCENESSE clinical data package for NDA submission
Wednesday - July 13, 2016 @ 10:30:37
FDA accepts SCENESSE clinical data package for NDA submission
Clinuvel prepares New Drug Application (NDA) for the treatment of erythropoietic protoporphyria (EPP) Executive summary: FDA concludes initial review of datasets on SCENESSE in EPP FDA deems datasets satisfactory for submission of New Drug Application Pre-NDA meeting with FDA to be scheduled to discuss timelines and procedure Clinuvel will request rolling review of NDA under Fast Track designation Clinuvel to request Priority Review to shorten review from 10 to 6 months Clinuvel Pharmaceuticals [ASX: CUV; Nasdaq International Designation ADR: CLVLY; Xetra-DAX: UR9] today announced that the US Food and Drug Administration (FDA) has concluded an initial review of Clinuvel's clinical
This is great news for the EPP community! We are on our way to approval of Scenesse/Afamelanotide. Be sure to register to attend the FDA meeting to join us in our continued efforts to show the FDA how important it is for them to approve this drug in the US.
*After you register, you will receive a confirmation email from the FDA with various questions to answer.*
PLEASE let the APF know you will be attending once you have finished registering online. We will be hosting a gathering for everyone the evening before the meeting. You may call the office at 866-APF-3635 or email us at porphyrus@porphyriafoundation.com.
*Please note that if you are unable to attend in person, there is an option for you to attend via webcast.*
"Remember.Research is the key to your cure!"
EPP KIT? Do You have One? Get yours today
Monday - July 11, 2016 @ 10:00:36
EPP Kit
To help families and doctors manage EPP
â?¢ $30.00 for US residents
Essential education produced exclusively for EPP patients. The Kit includes:
EPP brochure and education. Important background information about EPP, management strategies, photosensitivity, and ways to avoid attacks.
ER Information Form. At-a-glance sheet for ER personnel to record your essential admission information.
List of leading APF EPP specialists and their contact information.
Sun Precautions catalog, including medical solutions for sun sensitive people.
Samples of light-protecting sun block and accompanying product information.
Order by calling the APF Office: Toll free: 1.866.APF.3635.
Erythropoietic Protoporphyria (EPP) and X-Linked Protoporphyria (XLP)
Friday - July 8, 2016 @ 19:45:38
Erythropoietic Protoporphyria (EPP) and X-Linked Protoporphyria (XLP)
Erythropoietic Protoporphyria (EPP) or Protoporphyria
Erythropoietic Protoporphyria is characterized by abnormally elevated levels of protoporphyrin IX in erythrocytes (red blood cells) and plasma (the fluid portion of circulating blood), and by sensitivity to visible light that is usually noticed in early childhood and occurs throughout life. EPP can result either from mutations of the ferrochelatase gene (FECH), or less commonly the delta-aminolevulinic acid synthase-2 gene (ALAS2). When EPP is due to an ALAS2 mutation it is termed X-linked protoporphyria (XLP), because that gene is found on the X chromosome.
Protoporphyrin accumulates first in the bone marrow in EPP, and then in red blood cells, plasma and sometimes the liver. Protoporphyrin is excreted by the liver into the bile, after which it enters the intestine and is excreted in the feces. It is not soluble in water so is not excreted in the urine.
EPP is the third most common type of porphyria, and the most common in childhood. It causes very painful photosensitivity and can greatly impair quality of life. Delay in diagnosis is greater than with any other type of porphyria.
Swelling, burning, itching, and redness of the skin may appear during or after exposure to sunlight, including sunlight that passes through window glass. This can cause mild to severe burning pain on sun-exposed areas of the skin. Usually, these symptoms subside in 12 to 24 hours and heal without significant scarring. Blistering and scarring are characteristic of other types of cutaneous porphyria but are unusual in EPP. Skin manifestations generally begin early childhood and are more severe in the summer.
There is an increased risk of gallstones, which contain protoporphyrin. Excess protoporphyrin can also cause liver damage. Less than 5% of EPP patients severe liver damage and a condition caused protoporphyric hepatopathy that sometimes requires liver transplantation.
Diagnosis and Genetic Counseling
EPP should be suspected in anyone with non-blistering photosensitivity especially when it is prolonged and beginning in childhood. It is easy to make a diagnosis, or rule it out, once it is suspected.
The diagnosis of EPP is established by finding an abnormally high level of total erythrocyte protoporphyrin, and showing that this increase is mostly free protoporphyrin rather than zinc protoporphyrin. There is considerable confusion about which test to order. Sometimes laboratories have measured only zinc protoporphyrin and reported results incorrectly as protoporphyrin or free erythrocyte protoporphyrin (FEP). Laboratories that measure total erythrocyte protoporphyrin, free protoporphyrin and zinc protoporphyrin and report results reliably are:
Porphyria Laboratory and Center, University of Texas Medical Branch at Galveston, 1-409-772-4661
Mayo Medical Laboratories, 1-800-533-1710
ARUP Laboratories
Porphyrins are almost always elevated in plasma in EPP, but may be normal in mild cases. Fecal porphyrins may be normal or increased.
An experienced biochemical laboratory can usually distinguish between patients with EPP and XLP, because the former have much less zinc protoporphyrin in their erythrocytes. This can be explained because in the marrow the enzyme ferrochelatase not only normally makes heme (iron protoporphyrin) from protoporphyrin and iron, but can also make zinc protoporphyrin, especially when excess protoporphyrin is present or iron is deficient. However, this does not replace DNA studies.
Rarely, EPP develops in adults in the presence of a bone marrow disorder such as polycythemia vera, and is due to expansion of a clone of red blood cell precursors in the marrow that is deficient in ferrochelase.
DNA studies are important for confirming the diagnosis of EPP and XLP and for genetic counseling. This should be completed first in a person known to have the disease, and the information about the mutations in that individual used to guide testing of family members.
When EPP is due to a FECH mutation the inheritance is described as autosomal recessive. It is most common to find that one severe mutation is inherited from one parent and another weak mutation inherited from the other parent. The weak mutation is quite common in normal Caucasians, rare in Blacks and even more common in Japanese and Chinese populations. This mutation is sometime referred to as hypomorphic because it results in formation of a less than normal amount of ferrochelatase. But is does not cause EPP unless it is paired with a severe mutation. The severe mutation is characteristic for an EPP family and is present in all affected individuals. Carriers of the severe mutation are not affected because they do not have the weak mutation. Affected individuals and unaffected carriers can transmit the severe mutation to the next generation. Some of their children will have EPP if the other parent has a copy of the weak mutation. Rarely, the weak mutation is absent in an EPP family and two severe mutations are found, with at least one producing some ferrochelatase.
In XLP, mutations of the ALAS2 gene, which is found on the X chromosome, causes an increase in the production of the enzyme ALAS2 in the bone marrow. Several of these gain of function mutations have been described in different XLP families. In XLP protoporphyrin production exceeds that needed for heme and hemoglobin formation. Like hemophilia and other X linked genetic diseases, XLP is more common in men. Women have two X chromosomes and are usually not affected because they have a normal as well as a mutated ALAS2 gene. Men have only one X chromosome and will be affected if they inherit an ALAS2 mutation. Women with an ALAS2mutation will, on average, pass that mutation to half of their daughters (who will usually be unaffected carriers) and to half of their sons (who will be affected).
Treatment and Management
1. Sunlight protection
Protection from sunlight is the mainstay of management of EPP, and this is necessary throughout life. Disease severity and porphyrin levels in erythrocytes and plasma probably remain high and relatively constant throughout life in EPP. However, this has been little studied and more longitudinal observations are needed. Life style, employment, travel and recreation require adjustment in order to avoid painful reactions to sunlight and even from exposure to fluorescent lighting. For these reasons EPP can substantially affect quality of life.
Protective clothing, including broad-brimmed hats, long sleeves, gloves and trousers (rather than shorts), is beneficial. Several manufacturers specialize on clothing made of closely woven fabrics for people with photosensitivity.
2. Beta-Carotene (Lumitene Tishcon)
Beta-carotene is an over the counter product that was originally developed in a purified form as a drug for the treatment of EPP, and was shown to be effective by Dr. Micheline Mathews-Roth at Harvard University and others. The pharmaceutical grade formulation is now distributed by Tishcon as Lumitene, and can be ordered by calling 1-800-866-0978 or via the website www.epic4health.com. Other products are less standardized and reliable and are not recommended.
Beta-carotene provides protection by quenching reactive oxygen products that form when protoporphyrin is activated in the skin by light. It is important to take an amount that is adequate to be protective. For more information about Lumitene, including a recommended dosing schedule, please see the Lumitene section of this website.
3. Other considerations
In an occasional patient, protoporphyrin causes liver problems, so monitoring liver function is important. EPP patients should also not use any drug or anesthetic which causes cholestasis (slowing down bile flow), and should also avoid alcohol. Women should avoid medications containing estrogen (birth-control pills, hormone replacement therapy), and men should avoid testosterone supplements, as these substances can also have deleterious effects on the liver of a person with EPP.
Consult a specialist. Because EPP is a rare condition, most physicians are not knowledgeable about it. Contact The American Porphyria Foundation, 713-266-9617 for contact with an expert and to provide further information. A Medic Alert bracelet with instructions to contact a specialist if needed is a worthwhile precaution.
Yearly monitoring. Testing to include erythrocyte total protoporphyrin, plasma porphyrin, complete blood counts, ferritin and liver function tests should be done yearly. Porphyry levels are expected to be stable and liver tests to remain normal. EPP patients may have evidence of iron deficiency, and an iron supplement may be advisable if the serum ferritin is below about 20 ng/mL.
Vitamin D. Because they avoid sunlight, EPP patients are likely to be deficient in vitamin D. A vitamin D supplement with calcium is recommended for bone health.
Liver protection. It is important to avoid other causes of liver disease that might promote the development of liver complications from EPP. Patients should avoid alcohol and other substances that might damage the liver, including many herbal preparations, and be vaccinated for hepatitis A and B.
Surgical lights. Strong operating room lights can cause photosensitivity of the skin and even surfaces of internal organs. Flexible membrane filters, such as CL5-200-X from Madico Co., are available to cover surgical lights and offer some protection. This is especially important in EPP patients with liver failure, which causes even greater increases in protoporphyrin levels and photosensitivity.
Drugs. Drugs that are harmful in other porphyrias are not known to make EPP worse, but are best avoided as a precaution. This may include estrogens and other drugs that might reduce bile formation. A short course of a non-steroidal anti-inflammatory drug can provide some pain relief after an episode of photosensitivity, but can cause ulcerations of the digestive track especially with prolonged use.
Laser treatment. According to Dr. Roth, laser treatments for hair removal or eye surgery have not been a problem in EPP people. But the doctor should be made aware of the diagnosis, and that laser output between 400 and 650 nanometers might be harmful. Before hair removal treatment, the doctor may irradiate a small area of the skin to be treated for the length of time it will take to do the hair removal to ascertain if the patient would react within the period of time that a reaction to sunlight would be expected in that patient.
Children with EPP. Avoiding sunlight can be difficult for children with EPP who have less sunlight tolerance than their friends. Camp Discovery is an option for such children. It provides a week-long summer camping experience of fishing, boating, swimming, water skiing, arts and crafts, and just plain fun for young people with skin disorders, and is sponsored by the American Academy of Dermatology. Full scholarships, including transportation, are provided by the American Academy of Dermatology through generous donations of their members and other organizations. Members of the Academy are asked to recommend candidates for Camp Discovery, so ask your child's doctor about sending your child to Camp Discovery.
Disneyland and Disney World are responsive to people with sun sensitivity. They will provide a pass to enable you to enter attractions without waiting in line in the sun.
Disneyland Go to "Town Hall" and explain your problem with photosensitivity. You should bring a physician's letter with you as well as an APF brochure explaining the type of Porphyria you have. The staff will ask you some questions about your limitations (e.g., whether or not you can climb stairs) and how many are in your group. Next time you return, be sure to bring the old card with you, as it will only take about half as long to go through the process on your next trip.
Disney World Proceed to the "Guest Relations" office at any park (Magic Kingdom, EPCOT, etc.) and request the Special Assistance Pass.
Remember to bring a doctor's note and explanation of your condition, because it is not necessarily visible. People on duty may not be familiar with light sensitivity and its consequences.
Research Opportunities
Patients with EPP and XLP can participate in the research through the Porphyrias Consortium. The American Porphyria Foundation has information on what research protocols are currently open.
·
The Porphyrias Consortium is conducting a Longitudinal Study to better define the natural history of the disease. This study is currently open for enrollment of new patients.
·
The Porphyrias Consortium will be starting a pilot study soon on a drug that may lower porphyrin levels in EPP.
·
Clinuvel Pharmaceuticals is developing afamelanotide (Scenesse) for the treatment of EPP. This drug is given by injection and increases skin pigmentation. Another study of this drug is expected to open within the next year.
All patients with porphyria are encouraged to enter the Porphyrias Registry at the Porphyrias Consortium website. A link to this website is found on the website of the American Porphyria Foundation. Registration demonstrates to NIH that patients and their families think that research on porphyrias is important. You can also ask that one of the 6 porphyria center in the Consortium contact you.
Nichol Kirby invites you to a Patient Education Meeting in Indianapolis, IN
Thursday - July 7, 2016 @ 10:00:30
APF member Nichol Kirby invites you to a Patient Education Meeting in Indianapolis, IN! We hope you will take advantage of this opportunity to meet friends who share your experiences with porphyria. There will be a presentation and a Q & A session for all attendees.
Bring your friends and family! We can't wait to see you all there!
Porphyrias are rare disorders that affect mainly the skin or nervous system and may cause abdominal pain. These disorders are usually inherited, meaning they are caused by abnormalities in genes passed from parents to children. When a person has a porphyria, cells fail to change body chemicals called porphyrins and porphyrin precursors into heme, the substance that gives blood its red color. The body makes heme mainly in the bone marrow and liver. Bone marrow is the soft, spongelike tissue inside the bones; it makes stem cells that develop into one of the three types of blood cellsred blood cells, white blood cells, and platelets.
The process of making heme is called the heme biosynthetic pathway. One of eight enzymes controls each step of the process. The body has a problem making heme if any one of the enzymes is at a low level, also called a deficiency. Porphyrins and porphyrin precursors of heme then build up in the body and cause illness.
Heme is a red pigment composed of iron linked to a chemical called protoporphyrin. Heme has important functions in the body. The largest amounts of heme are in the form of hemoglobin, found in red blood cells and bone marrow. Hemoglobin carries oxygen from the lungs to all parts of the body. In the liver, heme is a component of proteins that break down hormones, medications, and other chemicals and keep liver cells functioning normally. Heme is an important part of nearly every cell in the body.
Each of the eight types of porphyria corresponds to low levels of a specific enzyme in the heme biosynthetic pathway. Experts often classify porphyrias as acute or cutaneous based on the symptoms a person experiences:
Acute porphyrias affect the nervous system. They occur rapidly and last only a short time.
Cutaneous porphyrias affect the skin.
Two types of acute porphyrias, hereditary coproporphyria and variegate porphyria, can also have cutaneous symptoms.
Experts also classify porphyrias as erythropoietic or hepatic:
In erythropoietic porphyrias, the body overproduces porphyrins, mainly in the bone marrow.
In hepatic porphyrias, the body overproduces porphyrins and porphyrin precursors, mainly in the liver.
Table 1 lists each type of porphyria, the deficient enzyme responsible for the disorder, and the main location of porphyrin buildup.
The exact rates of porphyria are unknown and vary around the world. For example, porphyria cutanea tarda is most common in the United States, and variegate porphyria is most common in South America.1
1Pierach CA. Porphyrias. In: Bope and Kellerman, eds. Conns Current Therapy 2012. 1st ed. Philadelphia, PA: Saunders; 2011: 847850.
Most porphyrias are inherited disorders. Scientists have identified genes for all eight enzymes in the heme biosynthetic pathway. Most porphyrias result from inheriting an abnormal gene, also called a gene mutation, from one parent. Some porphyrias, such as congenital erythropoietic porphyria, hepatoerythropoietic porphyria, and erythropoietic protoporphyria, occur when a person inherits two abnormal genes, one from each parent. The likeliness of a person passing the abnormal gene or genes to the next generation depends on the type of porphyria.
Porphyria cutanea tarda is usually an acquired disorder, meaning factors other than genes cause the enzyme deficiency. This type of porphyria can be triggered by
too much iron
use of alcohol or estrogen
smoking
chronic hepatitis Ca long-lasting liver disease that causes inflammation, or swelling, of the liver
HIVthe virus that causes AIDS
abnormal genes associated with hemochromatosisthe most common form of iron overload disease, which causes the body to absorb too much iron
For all types of porphyria, symptoms can be triggered by
Some people with porphyria-causing gene mutations have latent porphyria, meaning they have no symptoms of the disorder. Symptoms of cutaneous porphyrias include
oversensitivity to sunlight
blisters on exposed areas of the skin
itching and swelling on exposed areas of the skin
Symptoms of acute porphyrias include
pain in the abdomenthe area between the chest and hips
pain in the chest, limbs, or back
nausea and vomiting
constipationa condition in which an adult has fewer than three bowel movements a week or a child has fewer than two bowel movements a week, depending on the person
urinary retentionthe inability to empty the bladder completely
confusion
hallucinations
seizures and muscle weakness
Symptoms of acute porphyrias can develop over hours or days and last for days or weeks. These symptoms can come and go over time, while symptoms of cutaneous porphyrias tend to be more continuous. Porphyria symptoms can vary widely in severity.
A health care provider diagnoses porphyria with blood, urine, and stool tests. These tests take place at a health care providers office or a commercial facility. A blood test involves drawing blood and sending the sample to a lab for analysis. For urine and stool tests, the patient collects a sample of urine or stool in a special container. A health care provider tests the samples in the office or sends them to a lab for analysis. High levels of porphyrins or porphyrin precursors in blood, urine, or stool indicate porphyria. A health care provider may also recommend DNA testing of a blood sample to look for known gene mutations that cause porphyrias.
Treatment for porphyria depends on the type of porphyria the person has and the severity of the symptoms.
Acute Porphyrias
A health care provider treats acute porphyrias with heme or glucose loading to decrease the livers production of porphyrins and porphyrin precursors. A patient receives heme intravenously once a day for 4 days. Glucose loading involves giving a patient a glucose solution by mouth or intravenously. Heme is usually more effective and is the treatment of choice unless symptoms are mild. In rare instances, if symptoms are severe, a health care provider will recommend liver transplantation to treat acute porphyria. In liver transplantation, a surgeon removes a diseased or an injured liver and replaces it with a healthy, whole liver or a segment of a liver from another person, called a donor. A patient has liver transplantation surgery in a hospital under general anesthesia. Liver transplantation can cure liver failure. More information is provided in the NIDDK health topic, Liver Transplantation.
Cutaneous Porphyrias
The most important step a person can take to treat a cutaneous porphyria is to avoid sunlight as much as possible. Other cutaneous porphyrias are treated as follows:
Porphyria cutanea tarda. A health care provider treats porphyria cutanea tarda by removing factors that tend to activate the disease and by performing repeated therapeutic phlebotomies to reduce iron in the liver. Therapeutic phlebotomy is the removal of about a pint of blood from a vein in the arm. A technician performs the procedure at a blood donation center, such as a hospital, clinic, or bloodmobile. A patient does not require anesthesia. Another treatment approach is low-dose hydroxychloroquine tablets to reduce porphyrins in the liver.
Erythropoietic protoporphyria. People with erythropoietic protoporphyria may be given beta-carotene or cysteine to improve sunlight tolerance, though these medications do not lower porphyrin levels. Experts recommend hepatitis A and B vaccines and avoiding alcohol to prevent protoporphyric liver failure. A health care provider may use liver transplantation or a combination of medications to treat people who develop liver failure. Unfortunately, liver transplantation does not correct the primary defect, which is the continuous overproduction of protoporphyria by bone marrow. Successful bone marrow transplantations may successfully cure erythropoietic protoporphyria. A health care provider only considers bone marrow transplantation if the disease is severe and leading to secondary liver disease.
Congenital erythropoietic porphyria and hepatoerythropoietic porphyria. People with congenital erythropoietic porphyria or hepatoerythropoietic porphyria may need surgery to remove the spleen or blood transfusions to treat anemia. A surgeon removes the spleen in a hospital, and a patient receives general anesthesia. With a blood transfusion, a patient receives blood through an intravenous (IV) line inserted into a vein. A technician performs the procedure at a blood donation center, and a patient does not need anesthesia.
Secondary Porphyrinurias
Conditions called secondary porphyrinurias, such as disorders of the liver and bone marrow, as well as a number of drugs, chemicals, and toxins are often mistaken for porphyria because they lead to mild or moderate increases in porphyrin levels in the urine. Only highnot mild or moderatelevels of porphyrin or porphyrin precursors lead to a diagnosis of porphyria.
People with an acute porphyria should eat a diet with an average-to-high level of carbohydrates. The recommended dietary allowance for carbohydrates is 130 g per day for adults and children 1 year of age or older; pregnant and breastfeeding women need higher intakes.2 People should avoid limiting intake of carbohydrates and calories, even for short periods of time, as this type of dieting or fasting can trigger symptoms. People with an acute porphyria who want to lose weight should talk with their health care providers about diets they can follow to lose weight gradually.
People undergoing therapeutic phlebotomies should drink plenty of milk, water, or juice before and after each procedure.
A health care provider may recommend vitamin and mineral supplements for people with a cutaneous porphyria.
2National Research Council. Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids (Macronutrients). Washington, D.C.: The National Academies Press; 2005.
Porphyrias are rare disorders that affect mainly the skin or nervous system and may cause abdominal pain.
Each of the eight types of porphyria corresponds to low levels of a specific enzyme in the heme biosynthetic pathway.
The exact rates of porphyria are unknown and vary around the world. For example, porphyria cutanea tarda is most common in the United States, and variegate porphyria is most common in South America.
Most porphyrias result from inheriting an abnormal gene, also called a gene mutation, from one parent.
Porphyria cutanea tarda is usually an acquired disorder, meaning factors other than genes cause the enzyme deficiency.
Symptoms of cutaneous porphyrias include
oversensitivity to sunlight
blisters on exposed areas of the skin
itching and swelling on exposed areas of the skin
Symptoms of acute porphyrias include
pain in the abdomen
pain in the chest, limbs, or back
nausea and vomiting
constipation
urinary retention
confusion
hallucinations
seizures and muscle weakness
A health care provider diagnoses porphyria with blood, urine, and stool tests.
Treatment for porphyria depends on the type of porphyria the person has and the severity of the symptoms.
The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and other components of the National Institutes of Health (NIH) conduct and support research into many diseases and conditions.
What are clinical trials, and are they right for you? Clinical trials are part of clinical research and at the heart of all medical advances. Clinical trials look at new ways to prevent, detect, or treat disease. Researchers also use clinical trials to look at other aspects of care, such as improving the quality of life for people with chronic illnesses. Find out if clinical trials are right for youExternal NIH Link.
This information may contain content about medications and, when taken as prescribed, the conditions they treat. When prepared, this content included the most current information available. For updates or for questions about any medications, contact the U.S. Food and Drug Administration toll-free at 1-888-INFO-FDA (1-888-463-6332) or visitwww.fda.govExternal Link Disclaimer. Consult your health care provider for more information.
This content is provided as a service of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), part of the National Institutes of Health. The NIDDK translates and disseminates research findings through its clearinghouses and education programs to increase knowledge and understanding about health and disease among patients, health professionals, and the public. Content produced by the NIDDK is carefully reviewed by NIDDK scientists and other experts.
The NIDDK would like to thank: Herbert L. Bonkovsky, M.D., Carolinas Health Care System
This information is not copyrighted. The NIDDK encourages people to share this content freely.
Jared Ulmers vlog called Porphyria J Check him out
Monday - June 27, 2016 @ 10:00:00
APF Member, Jared Ulmer, has started a vlog on YouTube called Porphyria J. It's a vlog devoted to creating community and awareness around Porphyria, specifically EPP, and how he lives with it during the sunniest and hottest season of the year. The first episode of the 13 week series, called Summer Shade, aired last night.
Comment, Like, Share and Subscribe for new episodes every Tuesday!
Jared will be covering a spectrum of topics and we hope you join him on his journey.
A modest person is realistic. He recognizes that there are limits to his time, energy, and resources. So rather than trying to change everything at once, he makes improvement gradually.
Trying to reach all your goals at the same time is a sure way to reach none of them!
WHAT YOU CAN DO
Work on your habits in realistic increments. The following steps may help:
Create two master listsa list of good habits that you would like to build and a list of any bad habits that you need to get rid of. Do not limit yourself; on each list, write down as many as you can think of.
Prioritize the items on your lists, numbering them in the order of importance to you.
Choose a few habitseven just one or twofrom each list, and focus on those. Then move on to the next one or two habits on each of your lists.
Speed up the process by replacing a bad habit with a good one. For example, if your list of bad habits includes watching too much TV and your list of good habits includes keeping in touch with loved ones, you could resolve: Instead of immediately turning on the TV when I get home from work each day, I will contact a friend or a relative and catch up. Tips for the day
"Remember.Research is the key to your cure!"
How to Harness Your Habits
Wednesday - June 22, 2016 @ 10:00:01
How to Harness Your Habits
00:00
01:53
WHEN Austins alarm clock goes off, he is sleepy. But he immediately gets out of bed, puts on the exercise clothes he laid out the night before, and goes for a brief jogjust as he has three times a week for the past year.
Laurie just had a fight with her husband. Angry and frustrated, she storms into the kitchen, pulls out a bag of chocolate candies, and eats them alljust as she seems to do every time she is upset.
What do Austin and Laurie have in common? Whether they realize it or not, both have been affected by a powerful forcethe force of habit.
What about you? Are there good habits that you would like to build in your life? Perhaps your goal is to exercise regularly, to get more sleep, or to keep in closer touch with loved ones.
On the other hand, maybe you would like to break a bad habit, such as smoking cigarettes, eating too much junk food, or spending excessive time on the Internet.
Admittedly, it can be difficult to overcome a bad habit. In fact, it has been said that a bad habit is like a warm bed on a cold day: its easy to get into and hard to get out of!
So how can you harness your habits and make them work for you instead of against you? "Remember.Research is the key to your cure!"
Patient Meet in Philadelphia
Monday - June 20, 2016 @ 10:00:03
Over the weekend, Ariel Lager hosted a successful patient meeting in Philadelphia.
There was a great turnout and Dr. Manish Thapar gave a wonderful presentation about the porphyrias before welcoming questions from the group. Alnylam Pharmaceuticals also made a great presentation about their current research studies.
Jessica from the APF was able to attend the meeting and was very happy to meet so many people!
**There will be another Patient Meeting in Indianapolis soon! Stay tuned for more information!** Please let the APF know if you'd like to host a meeting in your area!
If you would like to receive the Apf E-News that contains information about Events, Research News, Research opportunities, and much much more please join the APF: http://www.porphyriafoundation.com/content/join-apf
Over the weekend, Ariel Lager hosted a successful patient meeting in Philadelphia.
There was a great turnout and Dr. Manish Thapar gave a wonderful presentation about the porphyrias before welcoming questions from the group. Alnylam Pharmaceuticals also made a great presentation about their current research studies.
Jessica from the APF was able to attend the meeting and was very happy to meet so many people!
**There will be another Patient Meeting in Indianapolis soon! Stay tuned for more information!**
Please let the APF know if you'd like to host a meeting in your area!
If you would like to receive the Apf E-News that contains information about Events, Research News, Research opportunities, and much much more please join the APF: http://www.porphyriafoundation.com/content/join-apf
"Remember.Research is the key to your cure!"
Erythropoietic Protoporphyria (EPP) and X-Linked Protoporphyria (XLP)
Friday - June 17, 2016 @ 10:00:22
Erythropoietic Protoporphyria (EPP) and X-Linked Protoporphyria (XLP)
Erythropoietic Protoporphyria (EPP) or Protoporphyria
Erythropoietic Protoporphyria is characterized by abnormally elevated levels of protoporphyrin IX in erythrocytes (red blood cells) and plasma (the fluid portion of circulating blood), and by sensitivity to visible light that is usually noticed in early childhood and occurs throughout life. EPP can result either from mutations of the ferrochelatase gene (FECH), or less commonly the delta-aminolevulinic acid synthase-2 gene (ALAS2). When EPP is due to an ALAS2 mutation it is termed X-linked protoporphyria (XLP), because that gene is found on the X chromosome.
Protoporphyrin accumulates first in the bone marrow in EPP, and then in red blood cells, plasma and sometimes the liver. Protoporphyrin is excreted by the liver into the bile, after which it enters the intestine and is excreted in the feces. It is not soluble in water so is not excreted in the urine.
EPP is the third most common type of porphyria, and the most common in childhood. It causes very painful photosensitivity and can greatly impair quality of life. Delay in diagnosis is greater than with any other type of porphyria.
Swelling, burning, itching, and redness of the skin may appear during or after exposure to sunlight, including sunlight that passes through window glass. This can cause mild to severe burning pain on sun-exposed areas of the skin. Usually, these symptoms subside in 12 to 24 hours and heal without significant scarring. Blistering and scarring are characteristic of other types of cutaneous porphyria but are unusual in EPP. Skin manifestations generally begin early childhood and are more severe in the summer.
There is an increased risk of gallstones, which contain protoporphyrin. Excess protoporphyrin can also cause liver damage. Less than 5% of EPP patients severe liver damage and a condition caused protoporphyric hepatopathy that sometimes requires liver transplantation.
Diagnosis and Genetic Counseling
EPP should be suspected in anyone with non-blistering photosensitivity especially when it is prolonged and beginning in childhood. It is easy to make a diagnosis, or rule it out, once it is suspected.
The diagnosis of EPP is established by finding an abnormally high level of total erythrocyte protoporphyrin, and showing that this increase is mostly free protoporphyrin rather than zinc protoporphyrin. There is considerable confusion about which test to order. Sometimes laboratories have measured only zinc protoporphyrin and reported results incorrectly as protoporphyrin or free erythrocyte protoporphyrin (FEP). Laboratories that measure total erythrocyte protoporphyrin, free protoporphyrin and zinc protoporphyrin and report results reliably are:
Porphyria Laboratory and Center, University of Texas Medical Branch at Galveston, 1-409-772-4661
Mayo Medical Laboratories, 1-800-533-1710
ARUP Laboratories
Porphyrins are almost always elevated in plasma in EPP, but may be normal in mild cases. Fecal porphyrins may be normal or increased.
An experienced biochemical laboratory can usually distinguish between patients with EPP and XLP, because the former have much less zinc protoporphyrin in their erythrocytes. This can be explained because in the marrow the enzyme ferrochelatase not only normally makes heme (iron protoporphyrin) from protoporphyrin and iron, but can also make zinc protoporphyrin, especially when excess protoporphyrin is present or iron is deficient. However, this does not replace DNA studies.
Rarely, EPP develops in adults in the presence of a bone marrow disorder such as polycythemia vera, and is due to expansion of a clone of red blood cell precursors in the marrow that is deficient in ferrochelase.
DNA studies are important for confirming the diagnosis of EPP and XLP and for genetic counseling. This should be completed first in a person known to have the disease, and the information about the mutations in that individual used to guide testing of family members.
When EPP is due to a FECH mutation the inheritance is described as autosomal recessive. It is most common to find that one severe mutation is inherited from one parent and another weak mutation inherited from the other parent. The weak mutation is quite common in normal Caucasians, rare in Blacks and even more common in Japanese and Chinese populations. This mutation is sometime referred to as hypomorphic because it results in formation of a less than normal amount of ferrochelatase. But is does not cause EPP unless it is paired with a severe mutation. The severe mutation is characteristic for an EPP family and is present in all affected individuals. Carriers of the severe mutation are not affected because they do not have the weak mutation. Affected individuals and unaffected carriers can transmit the severe mutation to the next generation. Some of their children will have EPP if the other parent has a copy of the weak mutation. Rarely, the weak mutation is absent in an EPP family and two severe mutations are found, with at least one producing some ferrochelatase.
In XLP, mutations of the ALAS2 gene, which is found on the X chromosome, causes an increase in the production of the enzyme ALAS2 in the bone marrow. Several of these gain of function mutations have been described in different XLP families. In XLP protoporphyrin production exceeds that needed for heme and hemoglobin formation. Like hemophilia and other X linked genetic diseases, XLP is more common in men. Women have two X chromosomes and are usually not affected because they have a normal as well as a mutated ALAS2 gene. Men have only one X chromosome and will be affected if they inherit an ALAS2 mutation. Women with an ALAS2mutation will, on average, pass that mutation to half of their daughters (who will usually be unaffected carriers) and to half of their sons (who will be affected).
Treatment and Management
1. Sunlight protection
Protection from sunlight is the mainstay of management of EPP, and this is necessary throughout life. Disease severity and porphyrin levels in erythrocytes and plasma probably remain high and relatively constant throughout life in EPP. However, this has been little studied and more longitudinal observations are needed. Life style, employment, travel and recreation require adjustment in order to avoid painful reactions to sunlight and even from exposure to fluorescent lighting. For these reasons EPP can substantially affect quality of life.
Protective clothing, including broad-brimmed hats, long sleeves, gloves and trousers (rather than shorts), is beneficial. Several manufacturers specialize on clothing made of closely woven fabrics for people with photosensitivity.
2. Beta-Carotene (Lumitene Tishcon)
Beta-carotene is an over the counter product that was originally developed in a purified form as a drug for the treatment of EPP, and was shown to be effective by Dr. Micheline Mathews-Roth at Harvard University and others. The pharmaceutical grade formulation is now distributed by Tishcon as Lumitene, and can be ordered by calling 1-800-866-0978 or via the website www.epic4health.com. Other products are less standardized and reliable and are not recommended.
Beta-carotene provides protection by quenching reactive oxygen products that form when protoporphyrin is activated in the skin by light. It is important to take an amount that is adequate to be protective. For more information about Lumitene, including a recommended dosing schedule, please see the Lumitene section of this website.
3. Other considerations
In an occasional patient, protoporphyrin causes liver problems, so monitoring liver function is important. EPP patients should also not use any drug or anesthetic which causes cholestasis (slowing down bile flow), and should also avoid alcohol. Women should avoid medications containing estrogen (birth-control pills, hormone replacement therapy), and men should avoid testosterone supplements, as these substances can also have deleterious effects on the liver of a person with EPP.
Consult a specialist. Because EPP is a rare condition, most physicians are not knowledgeable about it. Contact The American Porphyria Foundation, 713-266-9617 for contact with an expert and to provide further information. A Medic Alert bracelet with instructions to contact a specialist if needed is a worthwhile precaution.
Yearly monitoring. Testing to include erythrocyte total protoporphyrin, plasma porphyrin, complete blood counts, ferritin and liver function tests should be done yearly. Porphyry levels are expected to be stable and liver tests to remain normal. EPP patients may have evidence of iron deficiency, and an iron supplement may be advisable if the serum ferritin is below about 20 ng/mL.
Vitamin D. Because they avoid sunlight, EPP patients are likely to be deficient in vitamin D. A vitamin D supplement with calcium is recommended for bone health.
Liver protection. It is important to avoid other causes of liver disease that might promote the development of liver complications from EPP. Patients should avoid alcohol and other substances that might damage the liver, including many herbal preparations, and be vaccinated for hepatitis A and B.
Surgical lights. Strong operating room lights can cause photosensitivity of the skin and even surfaces of internal organs. Flexible membrane filters, such as CL5-200-X from Madico Co., are available to cover surgical lights and offer some protection. This is especially important in EPP patients with liver failure, which causes even greater increases in protoporphyrin levels and photosensitivity.
Drugs. Drugs that are harmful in other porphyrias are not known to make EPP worse, but are best avoided as a precaution. This may include estrogens and other drugs that might reduce bile formation. A short course of a non-steroidal anti-inflammatory drug can provide some pain relief after an episode of photosensitivity, but can cause ulcerations of the digestive track especially with prolonged use.
Laser treatment. According to Dr. Roth, laser treatments for hair removal or eye surgery have not been a problem in EPP people. But the doctor should be made aware of the diagnosis, and that laser output between 400 and 650 nanometers might be harmful. Before hair removal treatment, the doctor may irradiate a small area of the skin to be treated for the length of time it will take to do the hair removal to ascertain if the patient would react within the period of time that a reaction to sunlight would be expected in that patient.
Children with EPP. Avoiding sunlight can be difficult for children with EPP who have less sunlight tolerance than their friends. Camp Discovery is an option for such children. It provides a week-long summer camping experience of fishing, boating, swimming, water skiing, arts and crafts, and just plain fun for young people with skin disorders, and is sponsored by the American Academy of Dermatology. Full scholarships, including transportation, are provided by the American Academy of Dermatology through generous donations of their members and other organizations. Members of the Academy are asked to recommend candidates for Camp Discovery, so ask your child's doctor about sending your child to Camp Discovery.
Disneyland and Disney World are responsive to people with sun sensitivity. They will provide a pass to enable you to enter attractions without waiting in line in the sun.
Disneyland Go to "Town Hall" and explain your problem with photosensitivity. You should bring a physician's letter with you as well as an APF brochure explaining the type of Porphyria you have. The staff will ask you some questions about your limitations (e.g., whether or not you can climb stairs) and how many are in your group. Next time you return, be sure to bring the old card with you, as it will only take about half as long to go through the process on your next trip.
Disney World Proceed to the "Guest Relations" office at any park (Magic Kingdom, EPCOT, etc.) and request the Special Assistance Pass.
Remember to bring a doctor's note and explanation of your condition, because it is not necessarily visible. People on duty may not be familiar with light sensitivity and its consequences.
Research Opportunities
Patients with EPP and XLP can participate in the research through the Porphyrias Consortium. The American Porphyria Foundation has information on what research protocols are currently open.
·
The Porphyrias Consortium is conducting a Longitudinal Study to better define the natural history of the disease. This study is currently open for enrollment of new patients.
·
The Porphyrias Consortium will be starting a pilot study soon on a drug that may lower porphyrin levels in EPP.
·
Clinuvel Pharmaceuticals is developing afamelanotide (Scenesse) for the treatment of EPP. This drug is given by injection and increases skin pigmentation. Another study of this drug is expected to open within the next year.
All patients with porphyria are encouraged to enter the Porphyrias Registry at the Porphyrias Consortium website. A link to this website is found on the website of the American Porphyria Foundation. Registration demonstrates to NIH that patients and their families think that research on porphyrias is important. You can also ask that one of the 6 porphyria center in the Consortium contact you.
FDA HERE WE COME DONT GIVE UP NOW> MEETING IN OCT 2016
Synonyms of Hepatoerythropoietic Porphyria
Wednesday - June 15, 2016 @ 10:00:02
Hepatoerythropoietic Porphyria
NORD gratefully acknowledges Ashwani K Singal, MD, MSc, Division of Gastroenterology and Hepatology, University of Alabama at Birmingham, for assistance in the preparation of this report.
Synonyms of Hepatoerythropoietic Porphyria
autosomal recessive PCT
HEP
General Discussion
Summary
Hepatoerythropoietic porphyria (HEP) is an extremely rare genetic disorder characterized by deficiency of the enzyme, uroporphyrinogen decarboxylase. This deficiency is caused by mutations of both copies of a persons UROD gene, which means that the disorder is inherited as an autosomal recessive trait. Most affected individuals have a profound deficiency of this enzyme and onset of the disorder is usually during infancy or early childhood. However, some individuals may have a mild form that can go undiagnosed until adulthood. The childhood form of HEP is often associated with painful, blistering skin lesions that develop on sun-exposed skin (photosensitivity). Affected areas of skin can scar and become discolored. There may be risk of bacterial infection. Abnormal, excessive hair (hypertrichosis) on affected skin is also common. Mild anemia and abnormal enlargement of the liver and/or spleen (hepatosplenomegaly) have also been reported. Mild cases of HEP may go unrecognized until adulthood and can be clinically indistinguishable from porphyria cutanea tarda (PCT), a related disorder that may be acquired or occur in individuals with a mutation of one UROD gene (autosomal dominant inheritance). Cutaneous photosensitivity is generally much more severe in HEP than in PCT. NORD has a separate report on porphyria cutanea tarda.
Introduction
HEP belongs to a group of disorders known as the porphyrias. This group of at least seven disorders is characterized by abnormally high levels of porphyrins and porphyrin precursors due to deficiency of certain enzymes essential to the creation (synthesis) of heme, a part of hemoglobin and other hemoproteins. There are eight enzymes in the pathway for making heme and at least seven major forms of porphyria. The symptoms associated with the various forms of porphyria differ. It is important to note that people who have one type of porphyria do not develop any of the other types. Porphyrias are generally classified into two groups: the "hepatic" and "erythropoietic" types. Porphyrins and porphyrin precursors and related substances originate in excess amounts predominantly from the liver in the hepatic types and mostly from the bone marrow in the erythropoietic types. Porphyrias with skin manifestations are sometimes referred to as "cutaneous porphyrias". The term "acute porphyria" is used to describe porphyrias that can be associated with sudden attacks of pain and other neurological symptoms. HEP is a hepatic and cutaneous porphyria.
Signs & Symptoms
The symptoms and severity of HEP can vary from one person to another. Onset is usually within the first two years of life, but mild cases that go undiagnosed until adulthood have been reported. Although HEP is associated with specific, characteristic symptoms, several factors, including the small number of identified cases, make it difficult to establish the full range of associated symptoms of the disorder.
Severe cutaneous photosensitivity is usually the first sign. Affected infants may have extremely fragile skin that that can peel or blister on minimal impact is common. Reddening of the skin is common (erythema). Blistering skin lesions can develop on sun-exposed skin such as the hands and face. Photosensitivity can be severe and can cause scarring, erosion, and disfigurement. Bacterial infection of skin lesions can occur.
Abnormal, excessive hair growth (hypertrichosis) may also occur on sun-exposed skin. Affected skin may darken or lose color (hyper- or hypopigmentation). Small bumps with a distinct white head (milia) may also develop. Some affected individuals have teeth that are reddish-brown colored (erythrodontia).
Low levels of circulating red blood cells (anemia) may also occur. Anemia may be due to the premature destruction of red blood cells (hemolysis). Anemia associated with HEP may be mild or severe. Severe anemia may be associated with fatigue, pale skin, irregular heartbeat, chest pain, dizziness, and abnormally cold hands and feet. Some individuals may have an abnormally enlarged liver and/or spleen (hepatosplenomegaly).
Mild cases of HEP can go undiagnosed until adulthood. Overt photosensitivity may not be seen and mild skin damage can be mistaken for other conditions during childhood.
Causes
HEP is caused by mutations of both alleles of the UROD gene. Genes provide instructions for creating proteins that play a critical role in many functions of the body. When a mutation of a gene occurs, the protein product may be faulty, inefficient, or absent. Depending upon the functions of the particular protein, this can affect many organ systems of the body.
HEP is inherited as an autosomal recessive trait. Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Recessive genetic disorders occur when an individual inherits the same abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
Investigators have determined that the UROD gene is located on the short arm (p) of chromosome 1 (1p34.1). Chromosomes, which are present in the nucleus of human cells, carry the genetic information for each individual. Human body cells normally have 46 chromosomes. Pairs of human chromosomes are numbered from 1 through 22 and the sex chromosomes are designated X and Y. Males have one X and one Y chromosome and females have two X chromosomes. Each chromosome has a short arm designated p and a long arm designated q. Chromosomes are further sub-divided into many bands that are numbered. For example, chromosome 1p34.1 refers to band 34.1 on the short arm of chromosome 1. The numbered bands specify the location of the thousands of genes that are present on each chromosome.
The UROD gene creates (encodes) an enzyme known as uroporphyrinogen decarboxylase (UROD), which is the fifth enzyme in the heme biosynthetic pathway. In HEP, UROD enzyme activity is usually less than 10% its normal levels. Such low enzyme activity results in the abnormal accumulation of specific porphyrins and related chemicals in body, especially within the bone marrow, red blood cells, liver and skin. Symptoms develop because of this abnormal accumulation of porphyrins and related chemicals. For example when porphyrins accumulate in the skin, they absorb sunlight and enter an excited state (photoactivation). This abnormal activation results in the characteristic damage to the skin found in individuals with HEP. The liver removes porphyrins from the blood plasma and secretes it into the bile. When porphyrins accumulate in the liver, they can cause toxic damage to the liver.
Affected Populations
HEP is an extremely rare disorder that affects males and females in equal numbers. Approximately 40 cases have been reported in the medical literature. The exact incidence or prevalence of HEP in the general population is unknown.
Related Disorders
Symptoms of the following disorders can be similar to those of HEP. Comparisons may be useful for a differential diagnosis.
Congenital erythropoietic porphyria (CEP) is a rare inherited metabolic disorder resulting from the deficient function of the enzyme uroporphyrinogen III cosynthase (UROS), the fourth enzyme in the heme biosynthetic pathway. Due to the impaired function of this enzyme, excessive amounts of particular porphyrins accumulate, particularly in the bone marrow, plasma, red blood cells, urine, teeth, and bones. The major symptom of this disorder is hypersensitivity of the skin to sunlight and some types of artificial light, such as fluorescent lights (photosensitivity). After exposure to light, the photo-activated porphyrins in the skin cause bullae (blistering) and the fluid-filled sacs rupture, and the lesions often get infected. These infected lesions can lead to scarring, bone loss, and deformities. The hands, arms, and face are the most commonly affected areas. CEP is inherited as an autosomal recessive genetic disorder. Typically, there is no family history of the disease. Both parents are usually healthy, but each carries a defective gene that they can pass to their children. Affected offspring have two copies of the defective gene, one inherited from each parent. (For more information on this disorder, choose congenital erythropoietic porphyria as your search term in the Rare Disease Database.)
There are other conditions that may cause signs and symptoms that are similar to those seen in HEP. Such conditions include other cutaneous porphyrias, drug-induced photosensitivity, epidermolysis bullosa, various forms of lupus, and solar urticarial. (For more information on these disorders, choose the specific disorder name as your search term in the Rare Disease Database.)
Diagnosis
A diagnosis of HEP is based upon identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation and a variety of specialized tests. HEP may be considered in infants and children with chronic, blistering photosensitivity.
Clinical Testing and Workup
Screening tests can help diagnose HEP by measuring the levels of certain porphyrins in blood plasma, urine and red blood cells. These tests can help to differentiate the disorder from congenital erythropoietic porphyria by the different patterns of individual porphyrins and/or by demonstrating markedly decreased activity of the UROD enzyme. There is elevation of porphyrins in plasma, urine, and feces. Porphyrin patterns in HEP are similar to those seen in PCT with elevation of highly carboxylated porphyrins and isocoproporphyrins. In contrast to PCT, there are markedly increased levels of zinc protoporphyrin in red blood cells in HEP patients which is due to accumulation of pathway intermediates being metabolized to protoporphyrins.
Molecular genetic testing can confirm a diagnosis of HEP by detecting mutations in both UROD genes, but is available only on a clinical basis.
Standard Therapies
Treatment
The treatment of HEP is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, hematologists, dermatologists, hepatologists, and other healthcare professionals may need to systematically and comprehensively plan an affected childs treatment. Genetic counseling may benefit affected individuals and their families.
There is no specific, FDA-approved therapy for individuals with HEP. Because the disorder is so rare, most treatment information is based other forms of porphyria.
Avoidance of sunlight will benefit affected individuals and can include the use of clothing styles with long sleeves and pant legs, made with double layers of fabric or of light-exclusive fabrics, wide brimmed hats, gloves, and sunglasses. Topical sunscreens are generally ineffective, but certain tanning products with ingredients that increase pigmentation may be helpful. Affected individuals may also benefit from window tinting and the use of vinyl or films to cover the windows of their homes and cars.
Phlebotomies, which are used to treat individuals with PCT, are generally ineffective in individuals with HEP since elevated iron levels are not a feature of the disorder. Another treatment for PCT, the antimalarial drug chloroquine, was effective in at least one case reported in the medical literature.
Anemia may require treatment in some cases. Blood transfusions have been used to treat some individuals. Recombinant erythropoietin, which helps the body produce more red blood cells, was successfully used to treat severe anemia in an individual with HEP whose anemia was not associated with increased red cell destruction.
Investigational Therapies
Gene therapy is also being studied as another approach to therapy for individuals with genetic disorder associated with enzyme deficiency. In gene therapy, the defective gene present in a patient is replaced with a normal gene to enable the produce of the active enzyme and prevent the development and progression of the disease in question. Given the permanent transfer of the normal gene, which is able to produce active enzyme at all sites of disease, this form of therapy is theoretically most likely to lead to a cure. However, at this time, there remain some technical difficulties to resolve before gene therapy can be advocated as a viable alternative approach for genetic disorders like HEP.
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office: Toll-free: (800) 411-1222 TTY: (866) 411-1010 Email: prpl@cc.nih.gov
For information about clinical trials sponsored by private sources, in the main, contact: www.centerwatch.com
For more information about clinical trials conducted in Europe, contact: https://www.clinicaltrialsregister.eu/
HOORAY!! The APF, with your help, got an EPP meeting with the FDA! The meeting is scheduled for October 24, 2016 from 10am - 4pm (EDT) at the FDA. The FDA will be observing how many people are interested in this meeting so we need as many people as possible to attend. We MUST show them the extreme need for treatment. Great work, everyone! See below for more details! Register here: https://eppscientificworkshop.eventbrite.com
Dear Stakeholder,
On behalf of the Food and Drug Administration (FDA), we invite you to an upcoming public workshop on Erythropoietic Protoporphyria (EPP) to be held October 24, 2016 from 10am-4pm (EDT) at the FDA Campus in Silver Spring, Maryland. Specific details are outlined below.
The public workshop is intended to discuss how best to facilitate and expedite the development of safe and effective drug therapies to treat signs and symptoms related to EPP. FDA will provide information for and gain perspective from patients and patient advocacy organizations, health care providers, academic experts, and industry on disease symptoms and its impact on patients' daily life, patient experience with current treatment regimens for EPP, and various aspects of clinical development of products intended to treat EPP.
We're asking you to help make this workshop a success by encouraging patients, patient representatives, health care providers, academic experts and industry to participate either in-person or through the live webcast. FDA needs your help to make this workshop a success. Please direct stakeholders to the links below to register and to learn more about the workshop.
We look forward to this exciting workshop and hope to see you there. If you have any questions, please feel free to contact us at meghana.chalasani@fda.hhs.gov.
Use the link below to sign up: https://www.eventbrite.com/e/scientific-workshop-for-erythropoietic-protoporphyria-epp-registration-24971245668
"Remember.Research is the key to your c
ure!"
Dr. Bloomer Director of the Porphyria Center of University of AL of Birmingham speaks on ALAD
Wednesday - June 8, 2016 @ 12:48:01
ALAD Porphyria
NORD gratefully acknowledges Joseph R. Bloomer, MD, Professor of Medicine and Genetics, Director of the Liver Center, Director of the Porphyria Center, University of Alabama at Birmingham, for assistance in the preparation of this report.
Synonyms of ALAD Porphyria
ADP
ALAD deficiency
ALA-dehydratase deficient porphyria
delta-aminolevulinate dehydratase deficiency
Doss porphyria
porphyria of Doss
General Discussion
Summary
ALAD porphyria is a very rare genetic metabolic disease characterized by almost complete deficiency of the enzyme delta-aminolevulinic acid (ALA) dehydratase. Deficiency of this enzyme leads to the accumulation of the porphyrin precursor ALA, which can potentially result in a variety of symptoms. Symptoms vary from one person to another, but usually come from the neurological and gastrointestinal systems. This disease is inherited as an autosomal recessive disorder.
Introduction
ALAD porphyria is in the group of disorders known as the porphyrias. The porphyrias are characterized by abnormally high levels of porphyrins and porphyrin precursors in the body due to deficiencies of enzymes essential to the creation (synthesis) of heme, a part of hemoglobin. There are at least seven types of porphyria. The symptoms associated with the various types of porphyria differ. It is important to note that people who have one type of porphyria do not develop any of the other types. Porphyrias are generally classified into two groups: the "hepatic" and "erythropoietic" types. Porphyrins and related substances originate in excess amounts from the liver in the hepatic types, and mostly from the bone marrow in the erythropoietic types. ALAD porphyria is a hepatic form of porphyria.
Signs & Symptoms
The onset, severity and type of symptoms can vary greatly in individuals with a specific type of porphyria. This variation may depend on, in part, the amount of residual enzyme activity in each individual. Individuals with more significant enzyme deficiency may have more severe symptoms and earlier onset. Individuals with partial deficiency will have milder symptoms, and some individuals will not develop any symptoms (asymptomatic). It is important to note that affected individuals may not have all of the symptoms discussed below. Affected individuals should talk to their medical team about their specific case, associated symptoms and overall prognosis.
Individuals with ALAD porphyria may have bouts or attacks when symptoms are intense, which are referred to as neurovisceral or acute attacks. An attack may last for several weeks. During an attack, affected individuals may experience severe abdominal cramping or pain accompanied by vomiting and constipation. During infancy, gastrointestinal abnormalities may cause an affected child to fail to grow and gain weight as expected.
Several other neurological symptoms can occur during an acute attack due to problems with the nerves outside the central nervous system (peripheral neuropathy), resulting in numbness or tingling in the hands and feet, burning pain, sensitivity to touch, and a lack of coordination. In severe cases, the motor nerves are involved, resulting in loss or partial impairment of the ability to use voluntary muscles. ALAD porphyria can also be associated with psychological changes during an acute attack. In severe cases, loss of contact from reality (psychosis) has been reported.
Additional symptoms that occur during acute attacks include a rapid heartbeat (tachycardia), high blood pressure (hypertension), seizures, and breathing (respiratory) impairment.
Causes
ALAD porphyria is caused by mutations in the ALAD gene, and the disease is inherited as an autosomal recessive disorder. This means that both copies of the ALAD gene have a mutation. Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother.
Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual inherits one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
The ALAD gene contains instructions for creating the enzyme aminolevulinate dehydratase (ALAD), which is necessary for the production of heme. Heme is part of hemoglobin, which is the oxygen-carrying component of red blood cells. Heme is mainly produced in the bone marrow and the liver. Eight different enzymes are necessary for the creation of heme.
Mutations of the ALAD gene result in deficient levels ofporphobilinogen in the body, with accumulation of ALA, which causes the symptoms associated with ALAD porphyria.
A variety of different triggers have been identified that can precipitatean acute attack in individuals with ALAD porphyria. These triggers include alcohol, certain drugs, physical and psychological stress, infection, fasting (reduced caloric intake) and dehydration. The use of estrogen or progesterone is also suspect of triggering an acute attack.
Affected Populations
ALAD porphyria is an extremely rare disorder with few cases reported in the medical literature. Most cases have occurred in Europe. However, the disorder can potentially occur in any population. More males have been identified with ALAD porphyria than females in the medical literature, but the disorder affects probably males and females in equal numbers. Researchers suspect that some cases of ALAD porphyria go undiagnosed or misdiagnosed, making it difficult to estimate the true frequency of this disorder in the general population. The onset of ALAD porphyria is usually during infancy or childhood, but late-onset of the disorder (well into adult life) has also been reported.
Related Disorders
Symptoms of the following disorders can be similar to those of ALAD porphyria. Comparisons may be useful for a differential diagnosis.
Lead poisoning occurs when lead accumulates in the tissues of the body. This accumulation may occur slowly over months or years. The symptoms of lead poisoning vary depending upon the amount of lead exposure and the age of an affected individual. Lead poisoning can potentially cause behavioral changes such as irritability and sluggishness, and neurological abnormalities including poor coordination, convulsions, altered mental status, and disease of the nerves outside the central nervous system (peripheral neuropathy). Lead poisoning can also cause nonspecific symptoms including fever, joint pain, abdominal pain, nausea, vomiting and constipation. Lead inhibits the ALAD enzyme and can cause a clinical picture similar to ALAD porphyria. (For more information on this disorder, choose heavy metal poisoning as your search term in the Rare Disease Database.)
Tyrosinemia type I is a rare autosomal recessive genetic metabolic disorder that is caused by lack of the enzyme fumaryl acetoacetate hydrolase, which is needed for the break-down of the amino acid tyrosine. Failure to break down tyrosine leads to abnormal accumulation of tyrosine and its metabolites in the liver, potentially resulting in severe liver disease. Tyrosine may also accumulate in the kidneys and central nervous system. There is also accumulation of succinylacetone, which is a potent inhibitor of ALA dehydratase. Symptoms and physical findings associated with tyrosinemia type I appear in the first months of life and include failure to gain weight and grow at the expected rate (failure to thrive), fever, diarrhea, vomiting, an abnormally enlarged liver (hepatomegaly), and yellowing of the skin and the whites of the eyes (jaundice). Tyrosinemia type I may progress to severe liver disease, cirrhosis, and hepatocellular carcinoma if left untreated. Treatment with nitisinone and a low-tyrosine diet should begin as soon as possible after the diagnosis is confirmed. (For more information on this disorder, choose tyrosinemia as your search term in the Rare Disease Database.)
Acute intermittent porphyria (AIP) is a rare genetic metabolic disorder that is caused by deficiency of the enzyme porphobilinogen deaminase (PBG). This enzyme deficiency results in the accumulation of porphyrin precursors ALA and PBG in the body. The enzyme deficiency by itself is not sufficient to produce symptoms of the disease. Additional factors such as hormones, drugs and dietary changes trigger the appearance of symptoms. Symptoms of AIP may include severe abdominal pain, constipation, muscle weakness, rapid heartbeat (tachycardia), high blood pressure (hypertension), behavioral changes, seizures and disease of the nerves outside the central nervous system (peripheral neuropathy). Acute intermittent porphyria is inherited as an autosomal dominant trait. (For more information on this disorder, choose acute intermittent porphyria as your search term in the Rare Disease Database.)
Variegate porphyria is a rare genetic metabolic disorder that is caused by deficiency of the enzyme protoporphyrinogen oxidase. This leads to the accumulation of porphyrins and porphyrin precursors in the body, which, can potentially result in a variety of symptoms. Some affected individuals present with skin symptoms, some with neurological symptoms and some with both. Common skin (cutaneous) symptoms include hypersensitivity to sunlight with formation of blisters in sun-exposed areas. Common neurological symptoms include muscle weakness, muscle pain, convulsions, and behavioral issues. Affected individuals may also have gastrointestinal symptoms such as abdominal pain and vomiting. Variegate porphyria is caused by mutations in the PPOX gene. This genetic mutation is inherited as an autosomal dominant trait. Some individuals who inherit this mutation do not develop any symptoms. (For more information on this disorder, choose variegate porphyria as your search term in the Rare Disease Database.)
An acquired form of ALAD porphyria has also been described in which six diabetic patients with advanced renal disease developed a syndrome similar to acute intermittent porphyria after initiation of treatment with erythropoietin. The symptoms varied but resolved in all patients when erythropoietin was stopped, and reappeared in four patients when erythropoietin was restarted. In all of the patients, the enzyme ALA dehydratase was low. (For more information on this disorder, see Hedger et al. Compr Ther. 2006 Fall; 32(3): 163-71. PMID:17435269)
Diagnosis
A diagnosis of ALAD porphyria is made based upon identification of characteristic symptoms, a detailed patient history, and a thorough clinical evaluation and of specialized tests that can detect delta-aminolevulinic acid in the urine.
Molecular genetic testing can confirm a diagnosis of ALAD porphyria by identifying the characteristic genetic mutation that causes the disorder.
Standard Therapies
Treatment
The treatment of ALAD porphyria is directed toward the specific symptoms that are present in each individual. Because there have been so few cases of ALAD porphyria, there is only limited information on treatment for the disorder.
Avoidance of triggering factors such as alcohol, certain drugs, fasting, and low carbohydrate diets is recommended for affected individuals. The specific drugs that may need to be avoided in one person can differ from the drugs that need to be avoided in another. More information on these preventive measures and a list of drugs that may potentially need to be avoided are available from the American Porphyria Foundation (see Resources section of this report).
Two standard treatments for acute porphyrias in general are intravenous infusions of hemin and supplementation with glucose. However, these therapies have not been universally effective in treating individuals with ALAD porphyria.
Hemin is an orphan drug that has been approved by the Food and Drug Administration (FDA) for the treatment of acute porphyria. The drug known as Panhematin (hemin for injection) is usually given to treat an acute attack. The drug is manufactured by:
If hemin cannot be obtained quickly enough, glucose administration both orally and intravenously (which has similar effect to hemin) may be used to treat individuals with ALAD porphyria.
Additional drugs may be used to treat affected individuals during an acute attack including pain medications such as opiates, beta-adrenergic blocking agents such as propranolol to treat a rapid heartbeat, sedatives to calm nerves, drugs that reduce nausea and vomiting (anti-emetics) and anti-seizure medications (anti-convulsants). In addition, intravenous fluid replacement may be necessary during an acute attack to ensure that proper fluid and electrolyte levels are maintained.
Individuals with ALAD porphyria should carry Medic Alert bracelets or wallet cards. Genetic counseling may be of benefit for affected individuals and their families.
Investigational Therapies
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office: Tollfree: (800) 411-1222 TTY: (866) 411-1010 Email: prpl@cc.nih.gov
For information about clinical trials sponsored by private sources, contact: www.centerwatch.com
For information about clinical trials conducted in Europe, contact: https://www.clinicaltrialsregister.eu/
Ariel Lager will be hosting the next APF Patient Education Meeting in Philadelphia THIS Saturday, June 11th. There will be a presentation by porphyria expert, Dr. Manish Thapar. This is a great opportunity to have your questions answered by an expert and meet with others that share your experiences.
Bring your family and friends! Everyone is welcome! Please RSVP to Ariel Lager at 215-498-9002 or alager@ces-ltd.com to reserve your spot soon as possible!
Time
3:30-5:30 PM EST Location 1528 Walnut St, 21st Floor Philadelphia, PA 19102
*ANNOUNCEMENT* The "Moo've It In The Moonlight" Run in Burlington, NC has been POSTPONED. We will send out updated information as soon it is available.
SAVE THE DATES! Please RSVP to ATTEND
Monday - June 6, 2016 @ 15:32:12
Shawn Willis will be hosting the 2nd Annual "Moo've It In The Moonlight" Run in Burlington, NC on Friday, June 10 at 7:30 PM. The event is held in the evening to highlight the challenges of those with light sensitivity.There will be a 5K AND a 1 Mile Untimed Fun Run.The event is sponsored by Chick-fil-A and benefits the APF. The event was a huge success last year, so make sure to join us again this year!Visit the link below to RSVP!
Ariel Lager will be hosting the next APF Patient Education Meeting in Philadelphia, PA on Saturday, June 11th. The meeting will take place from 3:30-5:30 PM EST at the address shown below. There will be a presentation by porphyria expert, Dr. Manish Thapar.
Bring your family and friends! Everyone is welcome!
Clinuvel and FDA discuss EPP indication and regulatory pathways for SCENESSE
Monday, 05 October 2015 08:10
Clinuvel and FDA discuss EPP indication and regulatory pathways for SCENESSE
Type C meeting provides guidance for follow up and further discussions between FDA and Clinuvel
Leatherhead, UK and Melbourne, Australia, October 5, 2015
Clinuvel Pharmaceuticals Limited (ASX: CUV; ADR:CLVLY; XETRA:DAX) today announced that on September 30 it met in Silver Spring, USA, with the US Food and Drug Administrations (FDAs) Division for Dermatology and Dental Products (DDDP) and representatives of the Center of Drug Evaluation and Research (CDER). The objective of the meeting was to discuss the US regulatory review of SCENESSE (afamelanotide 16mg) to be made available to American erythropoietic protoporphyria (EPP) patients. In 2014 SCENESSE was granted marketing authorisation by the European Medicines Agency as a prophylactic photoprotective drug for adult EPP patients.
The DDDP stated that it was seeking a regulatory pathway to make SCENESSE available in the US, and the regulatory avenue of Accelerated Approval was suggested, pending FDAs review, analyses and further discussions on available photoprovocation and data on quality of life in EPP patients.
A discussion was held on the drugs benefits to patients who had received SCENESSE. Expert European and US porphyria clinicians were invited to share their experience in the treatment of EPP patients and the use of SCENESSE in their patients. Further discussions will be held with the DDDP following the review of photoprovocation and quality of life data.
- End -
References
Langendonk J et al (2015). Afamelanotide for Erythropoietic Protoporphyria. NEJM. 373(1):48-59.
Minder EI et al (2010). Patient-recorded outcome to assess therapeutic efficacy in protoporphyria-induced dermal phototoxicity: a proposal. Health Qual Life Outcomes. 8:60.
Rufener EA (1987). Erythropoietic protoporphyria: a study of its psychosocial aspects. Brit J Dermatol. 116:703-8.
EPP is characterised by phototoxicity (absolute intolerance to light) of the skin resulting in severe reactions, swelling, scarring and a state of distress. During phototoxic episodes patients experience longterm swelling of exposed body surfaces such as the face, hands and feet. A severe reaction triggered by exposure to light may result in hospitalisation. Patients do not respond to any analgesics or medication and following light exposure are typically unable to function. Due to the lifelong risk posed by light and UV, patients are often forced to lead an isolated indoor life deprived of normal activities.
Photoprovocation and Quality of Life data
Due to a genetic defect EPP patients are intolerant to light and sun. Photoprovocation is a standardised methodology of provoking limited EPP symptoms using specific controlled beams of light. In all five clinical studies of SCENESSE patients were subjected to photoprovocation under laboratory conditions.
Quality of life assessments utilise patient surveys to capture and analyse the impact of disease and treatment. Clinuvel has conducted quality of life surveys in all five EPP studies. Over the past eight years a disease-specific EPP Quality of Life questionnaire, developed by Clinuvel in association with expert EPP physicians, was used during clinical trials because other assessment tools were found to be unsuitable for evaluating the quality of life of EPP patients
Forward-Looking Statements
This release to the Australian Securities Exchange and to press contains forward-looking statements, including statements regarding future results, performance or achievements. These statements involve known and unknown risks, uncertainties and other factors which may cause Clinuvels actual results, performance or achievements to be materially different from any future results, performances or achievements expressed or implied by the forward-looking statements. These statements reflect our current views with respect to future events and are based on assumptions and subject to risks and uncertainties. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Some of the factors that could affect the forward-looking statements contained herein include: that the FDA may require additional studies beyond the studies planned for product candidates or may not provide regulatory clearances, including for SCENESSE; that the FDA may not provide regulatory approval for any use of SCENESSE or that the approval may be limited; that Clinuvel may never file an NDA for SCENESSE regulatory approval in the US; that the Company may not be able to access adequate capital to move its vitiligo programs forward; that the Company may not be able to retain its current pharmaceutical and biotechnology key personnel and knowhow for further development of its product candidates or may not reach favourable agreements with potential pricing and reimbursement agencies in Europe and the US.
This analysis describes the diagnosis and psychiatric treatment modalities of 6 patients (5 women, 1 man; mean age 28.5 years) of acute intermittent porphyria (AIP), who presented to the Psychiatry OPD over a period of one year. The mean number of episodes was 2.83. Premorbid personality traits, clinical presentation, urine colour and urinary porphobilinogen titre were recorded. Among the 6 patients, 4 had abdominal pain, 5 had autonomic instability, all 6 had mental symptoms, 3 had depression, 2 came in delirium, and 3 had an episode of seizure.
Keywords: Acute intermittent porphyria, neuropsychiatric manifestation of AIP, mental symptoms of AIP
Acute intermittent porphyria (AIP) is an autosomal dominant metabolic disorder that has various psychiatric manifestations. In AIP there is an abnormality in the haem biosynthetic pathway due to the deficiency of uroporphobilinogen I synthetase (porphobilinogen deaminase), which leads to excessive production of porphyrin precursors.1 The clinical features of AIP declare themselves at any stage from puberty onwards but mostly in the third decade of life. Penetrance of the gene responsible is incomplete, so that the disease often exists in a latent form and a family history may not be forthcoming.
The prevalence of AIP in the general adult population ranges between 1 and 8 per lakh.1,2 Tishler et al.3reported the overall prevalence of AIP in the psychiatric population to be 210 per lakh. Clinical symptoms are rare before puberty and the diagnosis of AIP is suggested by a triad of symptoms (abdominal pain, neuropathy and mental changes) and porphobilinogen in the urine. Urinary porphobilinogen is a moderately sensitive test. The diagnosis is confirmed by demonstrating a decreasing level of porphobilinogen in the erythrocytes. Autonomic lability is also highly suggestive of the diagnosis.4
Mental disorders accompany attacks in 24% to 80% of cases and psychiatric symptoms can dominate the picture. Psychiatric manifestations include hysteria, anxiety, depression, phobias, psychosis, organic disorders, agitation, delirium and altered consciousness ranging from somnolence to coma,5 and the most common neuropsychiatric manifestation reported are delirium and depression.
Although the diagnosis of porphyria is seldom made, it is still underdiagnosed due to lack of suspicion about the condition and lack of adequate investigative facilities. Six cases of porphyria are described who presented to the Department of Psychiatry, Assam Medical College, Dibrugarh, Assam at different occasions over a period of one year from January 1997 to December 1997. One patient who presented with classical symptoms is described in detail.
Ms YS, an 18-year-old tribal girl from a rural upper middle-class joint family with no history suggestive of psychiatric and metabolic disorders, had consulted many physicians and went to several psychiatric centres for 3 years since 1994. Her longitudinal history revealed that after attaining menarche at the age of 13 she used to have dysmenorrhoea, which subsided spontaneously. In 1994, at the age of 15 years, she was investigated for pain abdomen, and antitubercular treatment was started although there was no evidence of tuberculosis. The same treatment was continued by a physician on referral to Dibrugarh. She did not show any improvement; instead her condition deteriorated after discharge from hospital, because of which her medicines were discontinued. She however recovered spontaneously but continued to have episodes of pain abdomen, which made her very restless.
In March 1995, she was referred to a psychiatrist for vague somatic complaints. A diagnosis of conversion disorder was made. The patient never came for subsequent follow up, but on 26 December 1995 she reported with a history of poor sleep, crying spells, tingling of limbs, poor concentration and constipation. She was hospitalized and diagnosed as having adjustment disorder with depressed mood. Since her condition did not improve, she was taken to a private psychiatric nursing home at Guwahati on 24 February 1996, where EEG and CT scan of the brain were done and chlorpromazine and dothiapin was given at discharge. The patient was better for a month but again the same type of behaviour started and again she came to the Psychiatry OPD of Dibrugarh on 26 March 1996. She was admitted for her episodic abnormal behaviour, weeping spells, running away and suicidal attempt. Further, on the MSE she had obsessive rumination, pseudoauditory hallucination and depression. She was given clomipramine 175 mg, clonazepam 0.5 mg per day and 5 sessions of ECT. She recovered and remained well at home for 3 months with medicines although she still periodically suffered from abdominal pain.
In November 1996, while at home, she developed what appeared to be a generalized tonicclonic seizure complicating with paralysis and semi-stupor.
In December 1996, she developed florid symptoms of psychosis and was treated at the Imphal Medical College. She was discharged as a case of schizophrenia and long-acting neuroleptics were prescribed.
She came back to the Psychiatry OPD, Dibrugarh, in February 1997, with suicidal thoughts, sleeplessness, restlessness, feeling of sadness, lack of interest, swelling of abdomen with colicky pain lasting for brief periods and amenorrhoea of 2 months. On MSE, the patient was irritable, depressed and help-seeking; affect was la bella-type and occasionally labile; an elementary type of auditory hallucination was found. No physical abnormality was found except sinus tachycardia. Her Hamilton Depressive Rating Score was 27. At that stage, clinical AIP was considered and urine was tested for porphobilinogen, which was present in 1:80 titre. An incidental finding at ultrasonography was the presence of gallbladder stone.
Ms ND, a 35-year-old housewife and mother of two children, from a rural middle-class nuclear family, was referred on 16 August 1997 from the Medicine ward as a case of hysteria for her uncooperative behaviour and slurred speech. She was admitted to the Medicine ward on 12 August 1997 in a semiconscious state after repeated attacks of generalized tonicclonic seizures with no positive family history. Three months ago the patient had a similar seizure for which she was given phenytoin sodium, which caused marked confusion. Neurological assessment revealed the presence of peripheral neuropathy of all four limbs and residual XI and XII cranial nerve palsy as well as slurred speech. The patient was in delirium. Her high coloured urine raised a suspicion of porphyria. There was no pain abdomen. Urine showed porphobilinogen in 1:80 titre.
Ms YC, a 16-year-old girl, was referred by a physician on 6 September 1997 from a rural area with no suggestive family history or past history of any psychiatric illness. She presented with complaints of sleeplessness, irritability, aggressive outburst since she was treated with chloroquin for her fever 20 days back. On examination, she was afebrile and complained of episodic pain abdomen. On physical examination, no abnormality was detected except paroxysmal tachycardia. But on MSE she had fearfulness and panic attack; urine test for porphobilinogen was done on suspicion and it was detected in 1:80 titre.
Ms RB, a 34-year-old housewife and mother of two children, from an urban middle-class family, who was neurotic premorbidly and with no suggestive family history was referred to a psychiatrist in April 1996 by a general physician for her prolonged subjective unexplained multiple physical complaints including abdominal pain and constipation for the past 2 years. She was diagnosed as a case of somatoform disorder. During her several contacts with psychiatrists she had a fluctuating course that aggravated after imipramine, on her last visit in March 1997. She presented with bizarre somatic symptoms of delusional proportion with psychotic behaviour. Due to different presentation and for not showing improvements, porphyria was considered and urine for porphobilinogen was done and found positive in a 1:10 titre in consecutive samples. It was found negative during the remission period.
Ms AC, a 38-year-old housewife and mother of three children, from an urban middle-class family, was stable premorbidly with no suggestive family history but short-lasting psychosis of 3 months which subsided spontaneously, was referred by a physician as a case of hysteria to a psychiatrist with history of giddiness and loss of consciousness. On hospitalization the patient had atonic seizures and autonomic lability in the form of paroxysmal tachycardia and sweating and had anxiety with depression. Later, when she had severe pain abdomen with distension and vomiting, porphyria was considered and her consecutive urine samples were found positive for porphobilinogen in a 1:10 titre. On ultrasonography, cholecys-titis with cholelithiasis was detected.
Mr DC, a 32-year-old married man, cultivator from a rural area with no suggestive family history, was admitted with a history of forgetfulness, weakness, poor food intake, sleeplessness, deterioration in social functioning, abnormal behaviour and generalized tonicclonic seizures since one and a half months. He was never treated for seizures though these were present for 5 years. On starting with phenytoin, the patient gradually deteriorated and developed delirium and retention of urine. On noticing dark coloured urine, porphyria was suspected and porphobilinogen in urine was detected in 1:80 titre. Due to delayed diagnosis and treatment, the patient went into coma and died after 5 days.
Table 1 shows the age and gender distribution of the 6 patients (5 women and 1 men; average mean age 28.5 years). AIP is more frequent in women between second and fourth decades of life.6,7 Average mean age of onset was 27.6 years. Mean number of episodes was 2.83. Premorbid personality did not bear any particular personality traits.
Table 2 shows their clinical presentation in detail including the urine colour and urinary porphobilinogen titre. Among 6 patients, 4 had abdominal pain, 5 autonomic instability, and mental symptoms in all 6. Three patients had depression and 2 came in delirium. Depression and delirium are two neuropsychiatric manifestations that are most commonly reported.4 Among the 6 patients, 3 had seizures. The life-time prevalence of AIP associated with seizure was 2.2% of all those with known AIP and 5.1% of all those with manifest AIP.8
One patient (No. 6, DC) died while in coma due to AIP. According to Jeans et al.,9 mortality due to AIP was 3-fold compared to that in the general population during the past 50 years. The major cause of the increased mortality was the porphyric attack itself.
These patients were tested only qualitatively for porphobilinogen by using the Watson and Schwartz method. Quantitative tests for determination of 4-aminolevulinic acid (ALA) in urine could not be done due to lack of sophisticated laboratory facilities. But qualitative determination of porphobilinogen in the urine by Watson and Schwartz test is a simple and valuable screening test for the diagnosis of an acute attack of AIP. Hereditary coproporphyria (HCP) and variegated porphyria (VP) can be differentiated clinically from AIP by the presence of photosensitive cutaneous lesion and confirmed by titration of particular enzyme deficiency in erythrocytes, leukocytes and skin fibroblasts.
The Watson and Schwartz test is almost always positive during episodes of neuropsychiatric dysfunction but is positive only when the concentration of porphobilinogen in the urine is 3 to 5 times the upper limit of normal; as a consequence, both assays may be negative in latent cases and in patients in whom urinary excretion of porphobilinogen becomes normal following an acute attack. This however is by no means invariable and persistently elevated levels of porphobilinogen may occur. Caution is also required in certain febrile illness, in lead poisoning and in patients receiving phenothiazine drugs.10 Crimlisk11 reported that porphyria may be over-represented in psychiatric population, and negative screening test does not exclude the diagnosis. The diagnosis may be missed because it is not even considered or because of practical problems.
Depression is very common in porphyria but all textbooks are silent regarding safe antidepressants to be used. All standard textbooks mention imipramine and amitryptiline which can precipitate porphyria. There is uncertainty about newer antidepressants regarding their use in porphyria patients with depression, due to which the first patient could not be treated well. Clomipramine was given for a long time without signs of deterioration and unmodified ECT was safely used in one of the patients.
Again, regarding use of haloperidol and other anti-psychotic drugs besides phenothiazine nothing is mentioned in the textbooks. Theoretically we know that drugs that are metabolized by cytochrome P450 enzyme systems in the liver lead to the consumption of haem by cytochrome P450. This results in diminished levels of cellular concentration of haem, which in turn leads to depression of A-ALA synthetase with a corresponding increase in the rate of haem synthesis which may result in increased production of porphobilinogen in AIP patients.12
In conclusion, it is suggested that a list of safe antipsychotic and antidepressant drugs should be tried out and published to help in the treatment of porphyric patients with psychiatric manifestations.
1. Ackner B, Cooper JE, Gray CH, et al. Acute porphyriaa neuropsychiatric and biochemical study. J Psychosom Res. 962;6:124. [PubMed]
2. Goldberg A. Acute intermittent porphyria: A study of 50 cases. Q J Med. 1959;28:183209. [PubMed]
3. Tishler PV, Woodward B, O'Connor J, et al. High prevalence of intermittent acute porphyria in a psychiatric patient population. Am J Psychiatry. 1985;142:132036. [PubMed]
4. Paramala SJ, Malhotra S. Varied psychiatric manifestations of acute intermittent porphyria. Biol Psychiatry.1994;136:7447. [PubMed]
5. Burgovne K, Swartz R, Ananth J. Porphyria: Reexamination of psychiatric implications. Psychother Psychosom. 1995;64:12130. [PubMed]
6. Sugimara K. Acute intermittent porphyria. Nippon Rinsho. 1995;53:141821. [PubMed]
7. Bandr ES, Sanchez MP, Reynaog P. Acute intermittent porphyria at the hospital Arzobispo Loay-za of Lima (19831994)A report of 14 cases. Rev Gastroenterology Peru. 1994;14:20914. [PubMed]
8. Bylesjo I, Forsgren L, Lithner F, et al. Epidemiology and clinical characteristics of seizures in patients with acute intermittent porphyria. Epilepsia. 1996;37:2305. [PubMed]
9. Jeans JB, Savik K, Gross CR, et al. Mortality in patients with acute intermittent porphyria requiring hospitalization: A United States Case Series. Am J Med Genet. 1996;65:26973. [PubMed]
10. Reio LQ, Wetterberg L. False porphobilinogen reactions in the urine of mental patients. JAMA.1969;207:14850. [PubMed]
11. Crimlisk HL. The little imitatorPorphyria: A neuro-psychiatric disorder. J Neurol Neurosurg Psychiatry. 1997;62:31928. [PMC free article][PubMed]
12. Chatterjee MN, Rana S. Textbook of medical biochemistry. New Delhi: Jaypee Brothers; 1993. Porphyrias and porphyrinurias; pp. 30015.
"Remember.Research is the key to your cure!"
A Members Story of AIP
Friday - May 27, 2016 @ 10:00:24
Joe Hoss
Type of Porphyria:
Acute Intermittent Porphyria (AIP)
Joe Hosss Story AIP
I was about 30 when I had my first porphyria attack. It all started with severe abdominal pain, like the pain described by porphyria patients in the first 2016 APF newsletter. The pain was almost unbearable. They put me in the hospital and didnt know what was wrong. They began giving me medication but I was going downhill quickly. I called my mother to come to Birmingham, Alabama, where I was living, and she came down with porphyria symptoms around the same time. I was so confused and disoriented that I told my mother, much later, I dreamed that I was in a strait-jacket. But she told me that was no dream! Apparently I had gone into another patients room to use their shower, so they restrained me. My mother went into a coma while I was ill. I came to Jacksonville to be near other family after this. I had another 1-2 years of ineffective treatment and had a terrible backache. They gave me several over-the-counter drugs, but I was finally referred to a doctor that specialized in hand surgeries. He gave me a simple exercise and my back began to feel better quickly. This doctor recommended I begin swimming for exercise and this improved my situation tremendously. Swimming helped for about 10 years, from age 52-62, until I moved to another location that didnt have a pool I liked nearly as much.
I got married and shortly after, my attacks started again and I began having trouble digesting my food. A doctor gave me a new medication to alleviate this and it made me worse overall. The problem was actually solved with the transcendental meditation I started at age 33. I have been practicing since I was 33, and I am now 77. I also started yoga.
While my most severe pain occurred during my first attack, I continued to have porphyria pain and I wasnt diagnosed for several years after my first attack. My first attack was around the time when my mother went into a porphyria related coma. Later when they diagnosed my mother with Acute Intermittent Porphyria (AIP), they also tested me and I tested positive. I have tested positive on every 24-hour urine test Ive had in my whole life. My doctor prescribed a polycose powder for me to take in an attempt to keep my porphyria attacks at bay. I ingest local honey each day, along with peanut butter, and eat small meals every 4 hours, coupled with a traditional meal once/day.
Ever since my diagnosis, I have been a dedicated member of the APF.
"Remember.Research is the key to your cure!"
SCENESSE treatment in Europe
Thursday - May 26, 2016 @ 12:49:01
SCENESSE treatment in Europe
German Porphyria Expert Centres to start distribution of SCENESSE (afamelanotide 16mg) Clinuvel Pharmaceuticals Limited (ASX: CUV; XETRA-DAX: UR9; ADR: CLVLY) today announced an update on the company's post-authorisation distribution of SCENESSE (afamelanotide 16mg) across Europe for adult patients with the rare disorder erythropoietic protoporphyria (EPP).1 SCENESSE in EPP SCENESSE is the first approved treatment for EPP, a genetic disorder characterised by acute phototoxic reactions (anaphylactoid reactions and burns) and forced withdrawal from exposure to all forms of visible light. Clinuvel conducted clinical trials of SCENESSE in EPP from 2006 to 2013.
You may view the full Clinuvel announcement, along with others, at this address:
The APF would like to take this opportunity to remind you how important it is to continue our fight for this revolutionary drug to be approved in the USA.
PLEASE keep sending us your photos and stories. We will not stop submitting your stories to the FDA. Thank you for all you've done already!
"Remember.Research is the key to your cure!"
NORD and EPP: Micheline M. Mathews-Roth, MD, Associate Professor of Medicine, Harvard Medical School
Wednesday - May 25, 2016 @ 10:00:13
Erythropoietic Protoporphyria
NORD gratefully acknowledges Micheline M. Mathews-Roth, MD, Associate Professor of Medicine, Harvard Medical School, for assistance in the preparation of this report.
Synonyms of Erythropoietic Protoporphyria
EPP
Erythrohepatic Protoporphyria
Protoporphyria
General Discussion
Erythropoietic protoporphyria (EPP) is a rare inherited metabolic disorder characterized by a deficiency of the enzyme ferrochelatase (FECH). Due to abnormally low levels of this enzyme, excessive amounts of protoporphyrin accumulate in the bone marrow, blood plasma, and red blood cells. The major symptom of this disorder is hypersensitivity of the skin to sunlight and some types of artificial light, such as fluorescent lights (photosensitivity). After exposure to light, the skin may become itchy and red. Affected individuals may also experience a burning sensation on their skin. The hands, arms, and face are the most commonly affected areas. Some people with erythropoietic protoporphyria may also have complications related to liver and gallbladder function. Erythropoietic protoporphyria is inherited as an autosomal dominant genetic trait with poor penetrance.
Erythropoietic protoporphyria is one of a group of disorders known as the porphyrias. The porphyrias are all characterized by abnormally high levels of particular chemicals (porphyrins) in the body due to deficiencies of certain enzymes essential to the synthesis of hemoglobin. There are at least seven types of porphyria. The symptoms associated with the various types of porphyria differ, depending upon the specific enzyme that is deficient. It is important to note that people who have one type of porphyria do not develop any of the other types.
Signs & Symptoms
The most common symptom of erythropoietic protoporphyria is hypersensitivity of the skin to sunlight and some types of artificial light (photosensitivity), with pain, itching, and/or burning of the skin occurring after exposure to sunlight and occasionally to fluorescent light. Affected individuals may also exhibit abnormal accumulations of body fluid under affected areas (edema) and/or persistent redness or inflammation of the skin (erythema). In rare cases, affected areas of the skin may develop sac-like lesions (vesicles or bullae), scar, and/or become discolored (hyperpigmentation) if exposure to sunlight is prolonged. However, scarring and/or discoloring of the skin is uncommon and rarely severe. These affected areas of skin may become abnormally thick. In addition, in some cases, affected individuals may also exhibit malformations of the nails. The severity and degree of photosensitivity is different from case to case. Photosensitivity is often seen during infancy; however, in some cases, it may not occur until adolescence or adulthood.
In some affected individuals, the flow of bile through the gallbladder and bile ducts (biliary system) may be interrupted (cholestasis) causing gallstones (cholelithiasis) to form. In turn, such stones can cause obstruction and/or inflammation of the gallbladder (cholecystitis). Rarely, affected individuals may also develop liver damage that, in very severe cases, may lead to liver failure requiring transplantation.
Symptoms usually start in childhood but diagnosis is often delayed since blistering is not common and, because the porphyrins are insoluble, they usually escape detection on urinanalysis. The diagnosis is made upon finding increased levels of the protoporphyrin in the plasma or red blood cells.
Causes
Erythropoietic protoporphyria is a rare disorder inherited as an autosomal dominant genetic trait with poor penetrance. Human traits, including the classic genetic diseases, are the product of the interaction of two genes, one received from the father and one from the mother.
In dominant disorders, a single copy of the disease gene (received from either the mother or father) will be expressed dominating the other normal gene and resulting in the appearance of the disease. The risk of transmitting the disorder from affected parent to offspring is 50 percent for each pregnancy regardless of the sex of the resulting child. The risk is the same for each pregnancy.
The symptoms of erythropoietic protoporphyria develop due to excessive levels of a chemical called protoporphyrin that accumulates in certain tissues of the body (i.e., the plasma, red blood cells, and the liver). Excessive protoporphyrin levels occur as the result of abnormally low levels of the enzyme ferrochelatase (FECH).
There are several different allelic variants of erythropoietic protoporphyria. An allele is any of a series of two or more genes that may occupy the same position (locus) on a specific chromosome. Symptoms of these allelic variants of erythropoietic protoporphyria are predominantly the same; however, one type may be inherited as an autosomal recessive genetic trait.
The gene that is responsible for regulating the production of the enzyme ferrochelatase (FECH) has been located on the long arm of chromosome 18 (18q21.3). Chromosomes are found in the nucleus of all body cells. They carry the genetic characteristics of each individual. Pairs of human chromosomes are numbered from 1 through 22, with an unequal 23rd pair of X and Y chromosomes for males, and two X chromosomes for females. Each chromosome has a short arm designated as p and a long arm identified by the letter q.
Some people who have inherited this defective gene may have slightly elevated levels of protoporphyrin in the body but will not exhibit the symptoms of erythropoietic protoporphyria.
Affected Populations
Erythropoietic protoporphyria is a very rare inherited disorder that affects males and females in equal numbers. It is estimated that the disorder occurs in about 1 in about 74,300 individuals. The onset of symptoms affecting the skin usually occurs in infancy; however, in some cases, onset may not occur until adolescence or adulthood. More than 300 cases of EPP have been reported in the medical literature.
Related Disorders
Symptoms of the following disorders can be similar to those of EPP. Comparisons may be useful for a differential diagnosis:
There are several other types of porphyrias, all of which involve deficiencies of specific enzymes. Most of the symptoms of these porphyrias are not similar to the symptoms found in erythropoietic protoporphyria. Individuals with porphyria cutanea tarda and congenital erythropoietic porphyria may develop skin lesions; however, these lesions do not resemble the skin lesions found in EPP. It is important to note that individuals with one type of porphyria do not develop any of the other types. In addition, there are skin disorders characterized by hypersensitivity to artificial light and sunlight besides EPP, such as xeroderma pigmentosum and epidermolysis bullosa. The skin lesions in these disorders do not resemble the skin lesions in EPP. (For more information on these disorders, choose Porphyria and Epidermolysis Bullosa as your search terms in the Rare Disease Database.)
Xeroderma pigmentosum (XP) is a group of rare inherited skin disorders characterized by hypersensitivity of sunlight and some types of artificial light, with skin blistering occurring after such exposure. In some cases, pain and blistering may occur immediately after contact with sunlight or artificial light. Acute sunburn and persistent redness or inflammation of the skin (erythema) are also early symptoms of xeroderma pigmentosum. In most cases, these symptoms may be apparent immediately after birth or occur within the next three years. Other skin symptoms of xeroderma pigmentosum may include discolorations of the skin, weak and fragile skin, and/or scarring of the skin. Xeroderma pigmentosum also affects the eyes; the most common symptom being an extreme intolerance to light (photophobia). Additional symptoms may include some neurological impairments, short stature, an increased susceptibility to some forms of cancer (e.g., skin cancer). There are several types of xeroderma pigmentosum; in most cases, XP is inherited as an autosomal recessive genetic trait. (For more information on this disorder, choose Xeroderma Pigmentosum as your search terms in the Rare Disease Database).
Diagnosis
The diagnosis of erythropoietic protoporphyria (EPP) may be made by a thorough clinical evaluation, characteristic physical findings, and specialized laboratory tests. EPP is usually diagnosed during infancy or early childhood, due to characteristic skin symptoms. The diagnosis may be confirmed by testing the red blood cells (erythrocytes) for increased levels of protoporphyrin.
Standard Therapies
Treatment
Avoidance of sunlight will be of benefit to individuals with erythropoietic protoporphyria. The use of topical sunscreens, double layers of clothing, long sleeves, hats, and sunglasses will also benefit photosensitive individuals. Individuals with EPP may also benefit from window tinting or using vinyls or films to cover the windows in their car or house. Before tinting or shading car windows, affected individuals should check with their local Registry of Motor Vehicles to ensure that such measures do not violate any local codes.
In erythropoietic protoporphyria, a high potency form of oral beta-carotene (Lumitene, Tishcon) may be given to improve an affected individual's tolerance of sunlight. For more information on this treatment, contact the organizations listed at the end of this report (i.e., American Porphyria Foundation and the EPPREF) and Mr. George McShane of the Tishcon Corp. (1-800-848-8442). In some cases, the drug cholestyramine may be given to alleviate skin symptoms and lower the protoporphyrin levels in the body.
When iron deficiency is present, iron supplements may be given. A type of bile acid (chenodeoxycholic acid) may be prescribed to help the liver dispose of excess protoporphyrin, and activated charcoal or cholestyramine may be used to interrupt the circulation of protoporphyrin through the liver and intestines.
Estrogens and drugs that can impair bile flow should be given cautiously under the supervision of a physician. In addition, individuals with high levels of protoporphyrin in the plasma and red blood cells should be observed closely by a physician for possible liver malfunction that could eventually lead to liver failure.
Genetic counseling will be of benefit for affected individuals and their families. Other treatment is symptomatic and supportive.
Investigational Therapies
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. Government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Tollfree: (800) 411-1222
TTY: (866) 411-1010
Email: prpl@cc.nih.gov
For information about clinical trials sponsored by private sources, contact:
www.centerwatch.com
The orphan product L-Cysteine is being tested for the prevention and reduction of photosensitivity in erythropoietic protoporphyria. More research is needed to determine the long-term safety and effectiveness of this drug for the treatment EPP. For more information, contact:
Micheline M. Mathews-Roth, M.D.
Channing Laboratory
Harvard Medical School
181 Longwood Ave
Boston, MA 02115-5804
(617) 525-8249
Red blood cell transfusions have also been used to treat some people with EPP. In some affected individuals with severe liver disease, liver transplantations have been performed. Extreme caution should be used by physicians considering these treatment options; each particular case should be evaluated on its own merits.
Porphyria Facts from the NIH: Read and Share Dr.Porphyria Expert Herbert L. Bonkovsky, M.D., Carolinas Health Care System
Wednesday - June 15, 2016 @ 09:35:10
Porphyria Facts from the NIH: Read and Share Dr.Porphyria Expert Herbert L. Bonkovsky, M.D., Carolinas Health Care System
On this page: What are porphyrias? What is heme and what does it do? What are the types of porphyria? How common is porphyria? What causes porphyria? What are the symptoms of porphyria? How is porphyria diagnosed? How is porphyria treated? Eating, Diet, and Nutrition Points to Remember Clinical Trials What are porphyrias? Porphyrias are rare disorders that affect mainly the skin or nervous system and may cause abdominal pain. These disorders are usually inherited, meaning they are caused by abnormalities in genes passed from parents to children. When a person has a porphyria, cells fail to change body chemicals called porphyrins and porphyrin precursors into heme, the substance that gives blood its red color. The body makes heme mainly in the bone marrow and liver. Bone marrow is the soft, spongelike tissue inside the bones; it makes stem cells that develop into one of the three types of blood cellsred blood cells, white blood cells, and platelets.
The process of making heme is called the heme biosynthetic pathway. One of eight enzymes controls each step of the process. The body has a problem making heme if any one of the enzymes is at a low level, also called a deficiency. Porphyrins and porphyrin precursors of heme then build up in the body and cause illness.
[Top]
What is heme and what does it do? Heme is a red pigment composed of iron linked to a chemical called protoporphyrin. Heme has important functions in the body. The largest amounts of heme are in the form of hemoglobin, found in red blood cells and bone marrow. Hemoglobin carries oxygen from the lungs to all parts of the body. In the liver, heme is a component of proteins that break down hormones, medications, and other chemicals and keep liver cells functioning normally. Heme is an important part of nearly every cell in the body.
[Top]
What are the types of porphyria? Each of the eight types of porphyria corresponds to low levels of a specific enzyme in the heme biosynthetic pathway. Experts often classify porphyrias as acute or cutaneous based on the symptoms a person experiences:
Acute porphyrias affect the nervous system. They occur rapidly and last only a short time. Cutaneous porphyrias affect the skin. Two types of acute porphyrias, hereditary coproporphyria and variegate porphyria, can also have cutaneous symptoms.
Experts also classify porphyrias as erythropoietic or hepatic:
In erythropoietic porphyrias, the body overproduces porphyrins, mainly in the bone marrow. In hepatic porphyrias, the body overproduces porphyrins and porphyrin precursors, mainly in the liver. Table 1 lists each type of porphyria, the deficient enzyme responsible for the disorder, and the main location of porphyrin buildup.
Table 1. Types of porphyria
Type of Porphyria Deficient Enzyme Main Location of Porphyrin Buildup
congenital erythropoietic porphyria uroporphyrinogen III cosynthase bone marrow porphyria cutanea tarda uroporphyrinogen decarboxylase (~75% deficiency) liver hepatoerythropoietic porphyria uroporphyrinogen decarboxylase (~90% deficiency) bone marrow erythropoietic protoporphyria* ferrochelatase (~75% deficiency) bone marrow *Protoporphyria XLPP is a variant of erythropoietic protoporphyria.
[Top]
How common is porphyria? The exact rates of porphyria are unknown and vary around the world. For example, porphyria cutanea tarda is most common in the United States, and variegate porphyria is most common in South America.1
1Pierach CA. Porphyrias. In: Bope and Kellerman, eds. Conns Current Therapy 2012. 1st ed. Philadelphia, PA: Saunders; 2011: 847850.
[Top]
What causes porphyria? Most porphyrias are inherited disorders. Scientists have identified genes for all eight enzymes in the heme biosynthetic pathway. Most porphyrias result from inheriting an abnormal gene, also called a gene mutation, from one parent. Some porphyrias, such as congenital erythropoietic porphyria, hepatoerythropoietic porphyria, and erythropoietic protoporphyria, occur when a person inherits two abnormal genes, one from each parent. The likeliness of a person passing the abnormal gene or genes to the next generation depends on the type of porphyria.
Porphyria cutanea tarda is usually an acquired disorder, meaning factors other than genes cause the enzyme deficiency. This type of porphyria can be triggered by
too much iron use of alcohol or estrogen smoking chronic hepatitis Ca long-lasting liver disease that causes inflammation, or swelling, of the liver HIVthe virus that causes AIDS abnormal genes associated with hemochromatosisthe most common form of iron overload disease, which causes the body to absorb too much iron For all types of porphyria, symptoms can be triggered by
use of alcohol smoking use of certain medications or hormones exposure to sunlight stress dieting and fasting [Top]
What are the symptoms of porphyria? Some people with porphyria-causing gene mutations have latent porphyria, meaning they have no symptoms of the disorder. Symptoms of cutaneous porphyrias include
oversensitivity to sunlight blisters on exposed areas of the skin itching and swelling on exposed areas of the skin Symptoms of acute porphyrias include
pain in the abdomenthe area between the chest and hips pain in the chest, limbs, or back nausea and vomiting constipationa condition in which an adult has fewer than three bowel movements a week or a child has fewer than two bowel movements a week, depending on the person urinary retentionthe inability to empty the bladder completely confusion hallucinations seizures and muscle weakness Symptoms of acute porphyrias can develop over hours or days and last for days or weeks. These symptoms can come and go over time, while symptoms of cutaneous porphyrias tend to be more continuous. Porphyria symptoms can vary widely in severity.
[Top]
How is porphyria diagnosed? A health care provider diagnoses porphyria with blood, urine, and stool tests. These tests take place at a health care providers office or a commercial facility. A blood test involves drawing blood and sending the sample to a lab for analysis. For urine and stool tests, the patient collects a sample of urine or stool in a special container. A health care provider tests the samples in the office or sends them to a lab for analysis. High levels of porphyrins or porphyrin precursors in blood, urine, or stool indicate porphyria. A health care provider may also recommend DNA testing of a blood sample to look for known gene mutations that cause porphyrias.
[Top]
How is porphyria treated? Treatment for porphyria depends on the type of porphyria the person has and the severity of the symptoms.
Acute Porphyrias A health care provider treats acute porphyrias with heme or glucose loading to decrease the livers production of porphyrins and porphyrin precursors. A patient receives heme intravenously once a day for 4 days. Glucose loading involves giving a patient a glucose solution by mouth or intravenously. Heme is usually more effective and is the treatment of choice unless symptoms are mild. In rare instances, if symptoms are severe, a health care provider will recommend liver transplantation to treat acute porphyria. In liver transplantation, a surgeon removes a diseased or an injured liver and replaces it with a healthy, whole liver or a segment of a liver from another person, called a donor. A patient has liver transplantation surgery in a hospital under general anesthesia. Liver transplantation can cure liver failure. More information is provided in the NIDDK health topic, Liver Transplantation.
Cutaneous Porphyrias The most important step a person can take to treat a cutaneous porphyria is to avoid sunlight as much as possible. Other cutaneous porphyrias are treated as follows:
Porphyria cutanea tarda. A health care provider treats porphyria cutanea tarda by removing factors that tend to activate the disease and by performing repeated therapeutic phlebotomies to reduce iron in the liver. Therapeutic phlebotomy is the removal of about a pint of blood from a vein in the arm. A technician performs the procedure at a blood donation center, such as a hospital, clinic, or bloodmobile. A patient does not require anesthesia. Another treatment approach is low-dose hydroxychloroquine tablets to reduce porphyrins in the liver. Erythropoietic protoporphyria. People with erythropoietic protoporphyria may be given beta-carotene or cysteine to improve sunlight tolerance, though these medications do not lower porphyrin levels. Experts recommend hepatitis A and B vaccines and avoiding alcohol to prevent protoporphyric liver failure. A health care provider may use liver transplantation or a combination of medications to treat people who develop liver failure. Unfortunately, liver transplantation does not correct the primary defect, which is the continuous overproduction of protoporphyria by bone marrow. Successful bone marrow transplantations may successfully cure erythropoietic protoporphyria. A health care provider only considers bone marrow transplantation if the disease is severe and leading to secondary liver disease. Congenital erythropoietic porphyria and hepatoerythropoietic porphyria. People with congenital erythropoietic porphyria or hepatoerythropoietic porphyria may need surgery to remove the spleen or blood transfusions to treat anemia. A surgeon removes the spleen in a hospital, and a patient receives general anesthesia. With a blood transfusion, a patient receives blood through an intravenous (IV) line inserted into a vein. A technician performs the procedure at a blood donation center, and a patient does not need anesthesia. Secondary Porphyrinurias Conditions called secondary porphyrinurias, such as disorders of the liver and bone marrow, as well as a number of drugs, chemicals, and toxins are often mistaken for porphyria because they lead to mild or moderate increases in porphyrin levels in the urine. Only highnot mild or moderatelevels of porphyrin or porphyrin precursors lead to a diagnosis of porphyria.
[Top]
Eating, Diet, and Nutrition People with an acute porphyria should eat a diet with an average-to-high level of carbohydrates. The recommended dietary allowance for carbohydrates is 130 g per day for adults and children 1 year of age or older; pregnant and breastfeeding women need higher intakes.2 People should avoid limiting intake of carbohydrates and calories, even for short periods of time, as this type of dieting or fasting can trigger symptoms. People with an acute porphyria who want to lose weight should talk with their health care providers about diets they can follow to lose weight gradually.
People undergoing therapeutic phlebotomies should drink plenty of milk, water, or juice before and after each procedure.
A health care provider may recommend vitamin and mineral supplements for people with a cutaneous porphyria.
2National Research Council. Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids (Macronutrients). Washington, D.C.: The National Academies Press; 2005.
[Top]
Points to Remember Porphyrias are rare disorders that affect mainly the skin or nervous system and may cause abdominal pain. Each of the eight types of porphyria corresponds to low levels of a specific enzyme in the heme biosynthetic pathway. The exact rates of porphyria are unknown and vary around the world. For example, porphyria cutanea tarda is most common in the United States, and variegate porphyria is most common in South America. Most porphyrias result from inheriting an abnormal gene, also called a gene mutation, from one parent. Porphyria cutanea tarda is usually an acquired disorder, meaning factors other than genes cause the enzyme deficiency. Symptoms of cutaneous porphyrias include oversensitivity to sunlight blisters on exposed areas of the skin itching and swelling on exposed areas of the skin Symptoms of acute porphyrias include pain in the abdomen pain in the chest, limbs, or back nausea and vomiting constipation urinary retention confusion hallucinations seizures and muscle weakness A health care provider diagnoses porphyria with blood, urine, and stool tests. Treatment for porphyria depends on the type of porphyria the person has and the severity of the symptoms. [Top]
Clinical Trials The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and other components of the National Institutes of Health (NIH) conduct and support research into many diseases and conditions.
What are clinical trials, and are they right for you? Clinical trials are part of clinical research and at the heart of all medical advances. Clinical trials look at new ways to prevent, detect, or treat disease. Researchers also use clinical trials to look at other aspects of care, such as improving the quality of life for people with chronic illnesses. Find out if clinical trials are right for youExternal NIH Link.
What clinical trials are open? Clinical trials that are currently open and are recruiting can be viewed at www.ClinicalTrials.govExternal Link Disclaimer.
This information may contain content about medications and, when taken as prescribed, the conditions they treat. When prepared, this content included the most current information available. For updates or for questions about any medications, contact the U.S. Food and Drug Administration toll-free at 1-888-INFO-FDA (1-888-463-6332) or visit www.fda.govExternal Link Disclaimer. Consult your health care provider for more information.
[Top]
This content is provided as a service of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), part of the National Institutes of Health. The NIDDK translates and disseminates research findings through its clearinghouses and education programs to increase knowledge and understanding about health and disease among patients, health professionals, and the public. Content produced by the NIDDK is carefully reviewed by NIDDK scientists and other experts.
The NIDDK would like to thank: Herbert L. Bonkovsky, M.D., Carolinas Health Care System
This information is not copyrighted. The NIDDK encourages people to share this content freely.
The old adage, No Pain, No Gain, may work well in certain instances, but for porphyria, No Pain, No Gain is a terrible maxim. Most patients agree that porphyrias do not fit within the mainstream of painful conditions, like broken bones, labor pains, muscle spasms, etc. Porphyria pain is so severe that it is difficult to explain, but the people below have used their own extraordinary descriptions. After you read their quotes, you will understand why porphyria has gained a reputation as one of the most painful diseases.
Victor Mejias (EPP) It itches and burns so bad it makes me climb the walls.
Emily Melone (AIP) It is like charlie horses in my intestines.
Mike Boone (AIP) I want to unzip my skin and step out. My ribs feel like they are being fileted.
Karen Eubanks (AIP) I am being kicked with a steel toed boot.
Kim Stala (EPP) My hands feet and face are in boiling water for days while a knife is cutting and twisting my in-sides.
Monique Jacobs (HCP) My skin is akin to jellyfish stings while my belly burns like a washing machine full of lava.
Steve Stevens (AIP) My rib cage feels like the lava inside a volcano with lightning bolts going off.
Tara Cantley (AIP) My arm feels like someone cut off the circulation with a tie.
Lisa McFarland (EPP) It's like sticking your hands in a vat of hot frying oil with no relief in sight.
Stacy Hermann (AIP) I feel confused, angry, and full of deep despair. I cry at nothing at all.
Rachel Shinn Olstad (HCP) My daughter says it is like a knife stabbing her thigh over and over.
Debbie Rohn (AIP) My daughter says its like getting stung by thousands of bees and peroxide in her veins.
Deb Miller Gilbert (VP) My nerve endings are firing all at once and I do not get a break from it. My nerves are so overstimulated that a loving hug from my son is excruciating.
Tara Cantley (AIP) Someone is squeezing my arms and legs until they go numb and nerves feel as if they're burning.
Theresa Flagg Jurls (PCT) An inflated object with steel spikes caught behind my right ribcage.
Rachel Johnson Keith (AIP) Demons are chewing their way out of my gut and standing in a pile of fire ants.
Nicole Marie DiRoma (AIP) Severe burning torch in my stomach, being pregnant for your whole life and muscle pain like a 500lb person sitting on you and wont get off.
Rachel Ballew Campbell (AIP) It feels like someone is slicing your insides with a straight razor, while pouring hot lava through them. There really is no way to explain it.
Beverly Tuberville (AIP) Fire ants are all over.
Jennifer Taylor Nay (AIP) It feels like I am digesting glass shards.
Gregary Scott Allen Edwards (AIP) Its like being run over by a steam roller, shot by tasers repeatedly, and hav-ing all my life force batteries drained instantly.
Terri Witter (AIP) I have a tummy full of broken glass.
Mary Schloetter (AIP) My bones feel like rolling in glass shards and acid moving through my veins. Episodes are called 'attacks,' I agree as I'm standing alone on a battlefield being hit with guns, cannons and tasers.
Christine R. Baer (AIP) I have a knife through the abdomen, and my insides have jitters and dancing and moving.
Lisa Coleman-Vinson (AIP) Its like digesting huge, hot rocks with bone crushing pain.
Theresa Flagg Jurls (PCT) Steel spikes poking into my abdomen and ribcage, steel clamp gripped to pelvis.
Beth Nye (AIP) A 600lb person is sitting on my stomach and twisting it like a wet towel to remove all the water.
Nicki Theisen Maus (AIP) My insides feel like they're on fire!
Rogers Reyna (AIP) Its like aliens use my back as a trampoline while chewing their way through my gut. The nerve pain burn is fire ants having a festival in my arms and legs!
Patty Harris (AIP) Firing swords of hell with demons eating their way through your stomach back, legs, arms, and head. Lets face it, porphyria is burning hell.
Joe Mrsny (PCT) I look like I have leprosy because of the blisters breaking open. The itching is unbearable and causes scars and it usually comes at night so I am sleep deprived. The sun hurts. My finger nails fall off so I am unable to button my clothes. I just want to lay down and stay out of sight.
Whitney McCabe Stevens (HCP) Reality is, I can't accurately describe the pain of an attack because it is the most horrendous and overwhelming pain I have ever felt! On the pain scale of 1-10, it's 10,000. In my opinion, words simply can't accurately describe what we endure.
During a wonderful ceremony last night in Washington D.C., Desiree Lyon Howe, the Executive Director of the APF, was awarded the 2016 Rare Impact Award by the National Organization for Rare Disorders (NORD), which represents 23 million Americans with rare diseases. Desiree is grateful to receive this award and had a great time celebrating with her family, friends, and colleagues.Desiree credits the success of the APF to the members and the Scientific Advisory Board.
"Remember.Research is the key to your cure!"
VP FACTS
Monday - May 16, 2016 @ 10:00:18
Variegate porphyria (VP) is an autosomal dominant disorder that is a member of a family of disorders referred to as the porphyrias. Each disease in this family results from deficiencies in a specific enzyme involved in thebiosynthesis of heme (also called the porphyrin pathway). The term porphyria is derived from the Greek termporphura which means "purple pigment" in reference to the coloration of body fluids in patients suffering from a porphyria. The porphyrias are classified on the basis of the tissue that is the predominant site of accumulation of metabolic intermediates. These classifications are "hepatic" or "erythroid". Each disease is also further characterized as being acute or cutaneous dependent upon the major clinical features of the disease. VP results from defects in protoporphyrinogen oxidase. VP is classified as an acute hepatic porphyria. VP is also known by the names porphyria variegata, protocoproporphyria and South African genetic porphyria. The disease is quite common in South African whites (hence the related name of this disorder) with a frequency of 3 cases per 1000 persons. This high incidence has been traced to a founder couple who emigrated from Holland and were married in 1688.
The protoporphyrinogen oxidase gene (PPOX) is located on chromosome 1q22 spanning 8 kb and encompassing 194 exons that generate two alternatively spliced mRNAs, both of which encode the same 477 amino acid protein. Several different mutations have been identified in the PPOX gene resulting in VP. These mutations include missense, nonsense, and splice-site mutations as well as deletions and insertions. Except for the mutation found in the ancestors of the South African founder couple, most mutations in the PPOX gene are unique to each affected patient. The South African founder mutation is a missense mutation where tryptophan is substituted for arginine at amino acid 59 (R59W).
Reaction Catalyzed by Protoporphyrinogen Oxidase
Clinical Features of VP
The clinical manifestations of VP are highly similar to other acute porphyrias such as acute intermittent porphyria, AIP. The term "variegate" assigned to this particular porphyria relates to the fact that the disease can present with neurological manifestations, cutaneous photosensitivity or both. Common symptoms include cutaneous photosensitivity, abdominal pain, constipation, nausea, vomiting, hypertension, tachycardia, neuropathy and back pain. Disorientation and frank psychosis may be conspicuous features of VP. The symptoms of VP are rarely manifest prior to puberty. However, individuals homozygous for PPOX mutations exhibit severe clinical manifestations of VP at the outset of childhood. Clinical manifestation in VP becomes apparent in the event of an increased demand for hepatic heme production. This most often occurs due to drug exposure or some other precipitating factor such as an infection similar to the precipitating factors in AIP. If a drug is the cause of an attack its use should be discontinued immediately. Women using oral contraceptives are particularly susceptible to the cutaneous manifestations of VP. Most individuals (at least 75%) who inherit a PPOX mutation are asymptomatic throughout life.
Treatment of VP involves a high carbohydrate diet and during a severe attack an infusion of 10% glucose is highly recommended. Hemin arginate is used to treat acute attacks of variegate porphyria but is not effective in treating the cutaneous symptoms associated with the disorder.
APF 600 PAges sent to FDA Commissioner! Fight EPP
Friday - May 13, 2016 @ 10:30:43
Last week the APF sent this stack of over 600 PAGES (2 and 1/2 inches tall) of EPP pictures and stories to the FDA Commissioner! We have not slowed our efforts one bit and we will to continue to fight! Keep sending us your personal stories, letters, photos, etc We need to keep putting pressure on the FDA! Summer is HERE. It is unacceptable that we do not have approval of Afamelanotide/Scenesse and we will not stop until we have achieved our goal. Join the APF in fighting for this revolutionary treatment to be approved in the US!
"Remember.Research is the key to your cure!"
Part 2 of Care giving (Important)
Friday - May 13, 2016 @ 10:00:14
Part 2 of Care giving: The Caregiver also needs to stay well and balanced, physically, emotionally, mentally and spiritually. This may require that they need a break. Who would be able to come in and help? Can I trust just anyone? Asking for help or letting others help can take some of the pressure off you and allow you to take care of yourself. Family and friends often want to help but may not know how or what you need. Here are some tips for working with family and friends that have helped me and other porphyria patients:
Look for the areas that may need help, buy making a list.
Hold a regular family meeting to to keep everyone up to date. Very important to include the patient and let them express their wishes and concerns.
Ask family and friends when they are able to help and what jobs they think they can do. Be clear about what you need help with.
When you do hear back from them, note it on your list and make sure they haven taken care of what was needed.
Also show your appreciation to all family and friends for their continued support and help.
One thing that I have done with many people including myself is when sick and others help I keep a notebook by me who called, came to visit or help, and what he or she did, so that while it is fresh in my mind I can go back at a later time when i'm well to Thank them specifically for what they have done. But, What if I mess up?. Stay tuned for my next blog "Remember.Research is the key to your cure!"
The ALAD gene provides instructions for making an enzyme known as delta-aminolevulinate dehydratase. This enzyme is involved in the production of a molecule called heme. Heme is vital for all of the body's organs, although it is found mostly in the blood, bone marrow, and liver. Heme is an essential component of several iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood).
The production of heme is a multi-step process that requires eight different enzymes. Delta-aminolevulinate dehydratase is responsible for the second step in this process, which combines two molecules of delta-aminolevulinic acid (the product of the first step) to form a compound called porphobilinogen. In subsequent steps, four molecules of porphobilinogen are combined and then modified to produce heme.
At least 10 mutations in the ALAD gene can cause a rare form of porphyria called ALAD deficiency porphyria. Most of these mutations change single protein building blocks (amino acids) in delta-aminolevulinate dehydratase. These changes reduce the activity of the enzyme, allowing delta-aminolevulinic acid to build up to toxic levels in the body. This compound is formed during the normal process of heme production, but reduced activity of delta-aminolevulinate dehydratase allows it to accumulate to toxic levels. Very high levels of this compound can cause attacks of abdominal pain, vomiting, and other signs and symptoms of ALAD deficiency porphyria.
The porphyrias are a group of diseases caused by abnormalities in the production by the body of chemicals called porphyrins. Porphyrins are very important as they form haemoglobin that carries oxygen around the body in the red blood cells. The production of haemoglobin involves a chain of reactions in which one porphyrin is converted to another, and the porphyrias are diseases that result from genetic abnormalities in this process. If there is a block in the chain of reactions, there will be a build-up in the body of a particular porphyrin (which depends on where the block occurs), and porphyrins in high concentration are damaging to tissues. The problems caused by the different porphyrias relate to the particular porphyrin that accumulates.
What is congenital erythropoietic porphyria?
Congenital erythropoietic porphyria (CEP), also called Günthers disease after the doctor who first described it, is the rarest of the porphyrias. It is estimated that about 1 in every 2 3 million people are affected by CEP, which affects males and females equally, and occurs in all ethnic groups.
What causes congenital erythropoietic porphyria?
In CEP, there are high levels of a porphyrin called porphyrinogen in the bone marrow, blood and urine, which cause the symptoms and signs.
Is congenital erythropoietic porphyria hereditary?
Yes. The parents of someone with CEP have no symptoms of the condition themselves (and are called carriers of the condition), but each of them has a mutation in one of their genes. There is a 1 in 4 risk that each child born to 2 carriers will inherit the abnormal gene from both parents and thus develop the condition. This form of inheritance is called autosomal recessive.
What are the symptoms of congenital erythropoietic porphyria?
Individuals with CEP may not have all of the problems described in this leaflet as the severity of the condition varies. Usually, the disease shows itself soon after birth or in early childhood, but sometimes onset of disease is delayed until the teenage years or early adulthood.
Red urine is usually the first sign noticed in newborn babies with CEP. The intensity of the redness can vary from day to day.
The skin is very sensitive to light, especially direct sunlight, which may cause blisters or ulcers, which heal to leave scars. This most commonly happens at sun-exposed sites, for example the backs of the hands, the face, ears and scalp.
The eyes may also be sensitive to bright sunlight or artificial light, which can cause ulcers and scarring. With time, some patients lose their eyelashes, making their eyes easily irritated by small particles of dust and fibre.
The skin may take longer to heal after injury or blistering, and become infected.
Anaemia, which varies in severity, is common in CEP. Anaemia develops because porphyrin damages red blood cells, and causes tiredness, shortness of breath following minimal exertion, and paleness.
The spleen, which removes the damaged red blood cells, can gradually become bigger and cause worsening of the anaemia and a reduction in the number of platelets (the blood cells that help to form blood clots to stop bleeding) and white cells (the blood cells that fight infections) in the blood. This can lead to an increased risk of bleeding (such as repeated nose bleeds) and infections.
CEP can occasionally cause thinning of the bones (osteoporosis), which can lead to bone fractures following minor injury. What does congenital erythropoietic porphyria look like?
Repeated blisters and ulcers can cause extensive scarring in sun-exposed skin and permanent bald patches on the scalp. Some individuals may develop darkening of sun-exposed skin. Excess body hair may develop, especially on the face and backs of the hands.
How is CEP diagnosed?
CEP may be suspected in children (or rarely adults) who present with the problems described in this leaflet. The diagnosis is confirmed by measuring porphyrin levels in the blood, urine and faeces. These samples need to be protected from light until tested. A blood sample may also be taken to look for changes in the genes.
Testing for CEP in pregnancy is not offered routinely. However, CEP can be diagnosed in pregnancy in families where there is already a child with CEP. In this situation, cells taken from the fluid surrounding the baby in the womb (amniocentesis) or from the placenta, at 3 to 4 months into the pregnancy, are checked for the gene mutations causing CEP.
Can CEP be cured?
Currently, the only available cure for CEP is a bone marrow transplant (BMT). This involves transplanting healthy bone marrow from another person (the donor) to the person with CEP (the recipient). Following successful BMT, the symptoms of CEP such as photosensitivity and anaemia will improve. However, the scarring from previous damage to the skin is permanent.
For BMT to succeed, the bone marrow of the donor needs to be a good match with the recipient. BMT is a high-risk procedure, and is currently reserved for those severely-affected individuals who also have a bone marrow donor who is a close match.
Research is underway to cure CEP with gene therapy. This would involve correcting the abnormal gene in the affected person. It is hoped that this research will make good progress over the next decade.
For information about available treatments please visitthis pageon the website of the British Association of Dermatologists
Brought to you at this link: http://www.britishskinfoundation.org.uk/SkinInformation/AtoZofSkindisease/CongenitalErythropoieticPorphyria.aspx
"Remember.Research is the key to your cure!"
Being A Caregiver with Porphyria
Friday - May 6, 2016 @ 10:00:03
Being A Caregiver~ Part 1 What does a caregiver do? The caregiver is part of the health care team, which also includes the patient and the medical staff. Caregivers do many things:
Help feed, dress, and bathe the patient.
Make sure the patient eats and gets rest.
See that the patient takes medicines as they were told to.
Keep tracts of appointments.
Take care of insurance problems.
Drive the patient
Help with other family member's needs.
Talk to the health care team about how the patient is doing.
Help the patient live a normal a life as possible.
How to be a good caregiver: A good caregiver is often the one person who knows everything's that's going on with the patient. Don't be afraid to ask questions and take notes during Doctors visits. Learn who the members of the health care team are and know how to contact them. Let the person that has Porphyria make decisions. If the patient is making poor choices, talk to them about their choices. Then talk it over with the health care team and get their help. (Things like not taking medicines or not following the doctor's orders, may be poor choices that should not be ignored.) If you would like more information such as a Patient Kit or a Comprehensive Dr. Kit to be sent out, please contact the APF @ 1-866-APF-3635 Part 2 on Caregiver's help and text will be the next article:
"Remember.Research is the key to your cure!"
EPP Story of Maria Hensinger
Tuesday - May 3, 2016 @ 10:00:10
Maria Hensinger
Type of Porphyria:
Erythropoietic Protoporphyria (EPP)
I am a 38-year-old mother of two who lives right outside of Philadelphia. I was diagnosed with EPP at the age of two. My mother had noticed that I would get scabs on my nose when she took me out in the sun, so she took me to our family doctor who thought I had porphyria. Since this wasn't something anyone in our family had she got a second opinion at Children's Hospital of Philadelphia where they confirmed, through testing, that I did indeed have Erythropoietic Protoporphyria (EPP).
I have had outbreaks every year since then, though some years are better than others. My hands, nose and feet are the most affected. My symptoms are itching, burning, swelling, tingling, pain, sensitivity to different temperatures. Depending on the severity of the outbreak, my symptoms last from a day to a week. I limit my sun exposure to no more than an hour in full sun and about two hours covered up. I use tons of sunscreen and protective clothing (which I discovered only a few years ago).
I find most doctors do not know much about EPP. They make notations in my file but don't ask many questions about it. A good example would be that I took birth control pills for five years and just learned recently that I should not have taken them at all. I do wish I had learned that from one of my doctors. I find that European doctors seem to have a better understanding of my symptoms and how the disease works. I used to see a dermatologist for my outbreaks and when I was little I was tested periodically. There was never any information for me out there so I just tried to figure out my limits on my own and stopped seeing a doctor about the disease.*
When I became pregnant with my first child my husband and I spoke with genetic counselors at our hospital. They decided that we should test the baby right after birth to determine if he/she would have EPP. When my daughter was born we had her tested and she tested positive for it. When my son was born, he too tested positive for it. The doctors recommended that we retest them after they were two, as the tests they had as newborns could be a false positive.
My children are now 10 and seven and show no symptoms of EPP. A few years ago I had my son retested while he was having his normal checkup and his levels seemed to be a bit higher. At the recommendation of our pediatrician I consulted with the Chief of Hematology at duPont Children's Hospital in Delaware. The doctor contacted Dr. Micheline Mathews-Roth who indicated that my son could be a carrier but we could only know for sure with genetic testing. At this point, my husband and I decided that we would forgo it and let my son decide if he wanted it when he got older to determine whether or not his children may inherit EPP. I have no idea what my daughter's levels are, but will leave the genetic testing up to her if she wants it when she's older. My two sisters have been testedthey never showed any symptoms of EPP, and neither one is a carrier.
Through the testing process for my son, the doctor wanted me to get retested and so I did. The process of finding a lab that knew what was required was challenging as not all labs are equipped for doing the testing. It was reconfirmed that I had EPP but the doctor gave me all my results so I was able to see what my levels are, he also explained the genetic process and helped me really understand how it all happens. It is very lonely out there when you can't find any information on your illness or someone to relate to. A few years back I did a web search and found the APF website. What a thrill to find more information on my illness. I have been able to better understand my disease and educate my doctors on various parts of it. My family doctor has all the info I can find on EPP and is more than happy to make sure I have all my tests done for liver function, etc.
It's not easy living with EPP. My family has always made sacrifices to help me stay symptom-free. The outbreaks can make me delirious with pain and there isn't anything I can take for it. I try to keep my symptoms to myself but there are times when the pain is so great that I lose my temper and my patience. There have been many nights that I have had a bucket of cold water by my bed so I wouldn't have to get up to cool my hands or feet.
I try to stay out of the sun as much as possible but things like sun exposure through the car windows when I am driving can aggravate EPP. I now use gloves while driving in the spring and summer and a sun-protective scarf when I am a passenger. When I was younger I thought the hardest part was missing out on all the fun stuff myself, but after having kids I find that they have had to adapt to my limitations too and at times that makes me very sad. When they were babies and I had an outbreak, changing a diaper was a huge and very painful task and holding them was no better.
As my children got older and wanted to play outside they would protest when I would say "Mommy can't be out in the sun." Now they understand better and go with the flow of things, though at times I do need to remind them why we have to move inside. If anything I hope that I've instilled a sense of them taking care of their skin when they are going to be out in the sun. I also hope that the EPP stops with me and that their lifestyles will not be hindered once they have their own children.
*Editor's note. With more than 6,000 rare diseases in the world, the unfortunate fact is as Maria says: it is simply not possible for any doctor to be well versed in all of them. This means rare disease patients must rely on a combination of educating themselves and their doctors from reliable sources of medical information, and looking to expert consultation when it is available. The APF exists to provide all of these resources to patients and families dealing with all types porphyria. Please call our office to let us know how we can help you.
"Remember.Research is the key to your cure!"
What is Porphyria Read & Share
Tuesday - April 26, 2016 @ 10:30:05
What is Porphyria? Read & Share
Porphyria is a term that refers to a group of disordersthe porphyriasthat affect the nervous system or skin, or both. Each type of porphyria is due to the deficiency of one of the enzymes needed to make a substance in the body called heme. Enzymes are proteins that help chemical reactions happen in the body. Making heme involves a series of eight different enzymes, each acting in turn.
Heme is a red pigment composed of iron linked to a chemical called protoporphyrin. Heme has important functions in the body. The largest amounts of heme are in the blood and bone marrow in the form of hemoglobin within red blood cells. Hemoglobin gives blood its red color and carries oxygen from the lungs to all parts of the body. In the liver, heme is a component of proteins that have many functions, including breaking down hormones, drugs, and other chemicals and generating high-energy compounds that keep liver cells alive and functioning normally.
The body makes heme mainly in the bone marrow and the liver. The process of making heme is called the heme biosynthetic pathway. Each step of the process is controlled by one of eight enzymes. If any one of the enzymes is deficient, the process is disrupted. As a result, porphyrin or its precursorschemicals formed at earlier steps of the processmay build up in body tissues and cause illness.
What are the types of porphyria?
The table below lists each type of porphyria and the deficient enzyme responsible for the disorder. Porphyrias are often classified as acute or cutaneous. Acute types of porphyria affect the nervous system, whereas cutaneous types mainly affect the skin. Two forms of porphyriahereditary coproporphyria and variegate porphyriamay be either acute or cutaneous, or both.
TYPES OF PORPHYRIA
TYPE OF PORPHYRIA DEFICIENT ENZYME Acute Porphyrias ALAD porphyria delta-aminolevulinic acid dehydratase acute intermittent porphyria porphobilinogen deaminase hereditary coproporphyria coproporphyrinogen oxidase variegate porphyria protoporphyrinogen oxidase Cutaneous Porphyrias congenital erythropoietic porphyria uroporphyrinogen III cosynthase porphyria cutanea tarda uroporphyrinogen decarboxylase (~50% deficiency) hepatoerythropoietic porphyria uroporphyrinogen decarboxylase (~90% deficiency) hereditary coproporphyria coproporphyrinogen oxidase variegate porphyria protoporphyrinogen oxidase erythropoietic protoporphyria ferrochelatase The most common type of porphyria overall is porphyria cutanea tarda. In the United States, acute intermittent porphyria is the most common acute porphyria.
What causes porphyria?
Most porphyrias are inherited disorders, meaning they are caused by abnormalities in genes passed from parents to children. Scientists have identified the genes for all eight enzymes in the heme pathway. Some forms of porphyria result from inheriting an abnormal gene from one parent. Other forms are due to inheriting two abnormal genesone from each parent. The risk that members of an affected family will have the disease or transmit it to their children depends on the type of porphyria.
One type of porphyriaporphyria cutanea tardais most often an acquired disorder. It occurs when factors other than genes cause an enzyme deficiency in the liver.
Porphyria can be triggered by
drugs such as barbiturates, tranquilizers, birth control pills, and sedatives chemicals fasting smoking drinking alcohol, especially heavy drinking infections excess iron in the body emotional and physical stress menstrual hormones exposure to the sun What are the symptoms of porphyria?
People with cutaneous forms of porphyria develop blisters, itching, and swelling of their skin when it is exposed to sunlight. Symptoms of acute forms of porphyria include pain in the abdomen, chest, limbs, or back; numbness, tingling, paralysis, or cramping; vomiting; constipation; and personality changes or mental disorders. Acute attacks of porphyria can develop over hours or days and last for days or weeks.
Symptoms can vary widely in severity. Some people with gene mutations that can cause porphyria have no signs or symptoms of the disorder. These people are said to have latent porphyria.
How is porphyria diagnosed?
Doctors diagnose porphyria using blood, urine, and stool tests. Interpreting test results can be complex, and initial tests may be followed by further testing to confirm the diagnosis. Diagnosis may be delayed because the symptoms of porphyria are similar to symptoms of other disorders.
How is porphyria treated?
Each type of porphyria is treated differently. Treatment may involve avoiding triggers, receiving heme through a vein, taking medicines to relieve symptoms, or having blood drawn to reduce iron in the body. People who have severe attacks may need to be hospitalized.
Points to Remember
Porphyria is a group of disordersthe porphyriasthat affect the nervous system or skin, or both. Each type of porphyria results from a deficiency of one of the enzymes needed to make heme. Most porphyrias are inherited disorders, but porphyria cutanea tarda is usually an acquired disorder. Cutaneous types of porphyria affect the skin, causing symptoms such as blistering, itching, and swelling. Acute types of porphyria affect the nervous system, causing symptoms such as abdominal pain, vomiting, numbness, and mental disorders. Each type of porphyria is treated differently.
"Remember.Research is the key to your cure!" American Porphyria Foundation www.porphyriafoundation.org 4900 Woodway Dr. Suite 780 Houston, TX 77056
The APF would like to extend a huge THANK YOU to everyone who participated in making National Porphyria Awareness Week such a huge success this year. With your help, the APF reached over 50,000 people via social media! The APF mailed out MANY packets of brochures and other educational materials for distribution.
In addition to passing out educational materials, APF members engaged in other activities to promote porphyria awareness. On the first day of Awareness Week, Louise Schlosser hosted a very successful Patient Education Meeting in California. Some members set up tables outside of businesses during peak hours to educate the public. Nicholas Guanciale set up a porphyria table at his job. One member created a porphyria flyer to pass out at her upcoming family reunion. Other members sold porphyria-related items or created posts on Facebook. Another member even planted a garden spelling out EPP in honor of Erythropoietic Protoporphyria, as you saw featured in last week's enews!
Regardless of what you chose to do, the APF is deeply appreciative of your efforts and we hope you all enjoyed participating! We are already looking forward to next year!
My name is Allison Linner and I am seventeen. I came into this world on a cold, rainy day in early October, but my Dad said that the moment I was born, the sun peeked through the clouds, thus my nickname, Sunshine.
For some reason, my parents always knew there was something wrong with me. My life was dotted with strange symptoms, including lots of pain and lots of doctors for a girl my age. Each doctor diagnosed me with something else. When I hit puberty, things worsened. I had terrible episodes during which I would be doubled-over in front of the toilet, wanting to be unconscious because of the pain, but unwilling because I knew if I passed out I might choke on my own vomit. The toilet even became my pew. I fought to stay conscious while these other things were occurring. I could feel that my heartbeat was weak and irregular. Another strange symptom was that I was extraordinarily photosensitive. When ill, I would lie on my bed with my eyes shut, a wet washcloth over my eyes, the blinds closed tight, and I would still be irritated by light. This is one reason I thought I was just nuts. I even had times when I experienced paranoia and hallucinations. I would cry and pray hard. I wondered how my nickname could ever be Sunshine.
One of my most recent episodes was very hard. It was this past summer, and I decided to be the chaperone at an outdoor pool party my sister was attending. When I got out of the pool, one mom touched my face and said: Oh my Goshyoure so pale. I responded Yeah, I get that a lot. When we got back in the pool, I suddenly felt very ill and with Gods help I was able to get home and then get to the doctor.
I questioned him, In your professional opinion, do you think these symptoms that all of my different doctors have diagnosed over the years, are intertwined in some way? He said, No, there is no such disease that has all of your symptoms. But there had to be , I thought. I knew in my heart that each symptom was a puzzle piece and I realized that my family and I would have to play large roles in putting it together.
One day, my Dad and I talked a lot about what was happening. He promised me that I would find out what was wrong with me, and what was wrong with us, since we shared similar symptoms. At the end of our discussion, he jokingly said that maybe because we were so sensitive to light, we were vampires. He also said that there actually was a disease on which the legends were most likely founded. I thought I'd look it up, just for fun. Reading the list of symptoms was like reading the story of my life. It was actually a very spiritual experience, it felt like reading the scriptures. A few days later, I went to the doctor and asked him if he could order the tests. He said Sure, why not? While waiting on the obscure test results, I performed at home the '24-hour urine test.' Within a few hours, my urine was a deep purple-red hue. A few short days later, the tests came back positive for Hereditary Coproporphyria (HCP.)
This is a hard disease to have, there is no doubt about it. I know what pain is. But every day, I thank our Father in Heaven that I finally know what ails me; and that Im not crazy. Ive learned to cope with it daily. When I do have flare-ups that is probably when Im closer to God than any other time. It is so humbling. I am actually grateful for my disease because it does so much to compel me to be a better person.
When I turned 17 on October 10, my family gave me a card that has a cartoon sun smiling on it and it reads: Happy, Bright, and Lots of Fun. Inside, each member of my family wrote a little note to me. In my Dads message he wrote, You dont need the sun because you carry so much light inside you wherever you go. I guess Im still Sunshine after all.
Did you Know:
Hereditary coproporphyria (HCP)
HCP is similar to AIP, but the symptoms are typically milder. HCP is caused by a deficiency of coproporphyrinogen oxidase due to mutations in a gene by the same name at 3q12. The greatest difference between HCP and AIP is that people with HCP may have some skin sensitivity to sunlight. However, extensive damage to the skin is rarely seen.
"Remember.Research is the key to your cure!"
APF Awareneess Story Read Share Sandra Ihring VP
Friday - April 22, 2016 @ 11:10:14
Sandra Ihring
Variegate Porphyria (VP)
I come from a Central Oregon town with the population of about 1100 people. I moved back here to Central Oregon, where I was raised after 40 years of being away. I took my grandson to a local ER in Redmond, OR (population about 26,000) after an accident where he hurt his foot which needed to be checked out. I was making small talk with him in the waiting room and told him of some new information about porphyria I had learned. A woman in the waiting room overheard our conversation. So, as it goes in small towns; we started talking. She told me that she had porphyria also.
She was called in to see the triage nurse but her husband remained and we visited. I exchanged contact information with him and we briefly traded stories. She came back in the room shortly. My first question after finding out what kind of porphyria she had (AIP), I asked her how she was diagnosed; as I knew of my own 'war stories' of finally finding the correct diagnosis.
Her story was similar to mine and other porphyria patients, being brushed off by many regular doctors who do/did not understand the cluster of symptoms or level of pain involved during a porphyria attack. If your symptoms don't exactly fit a text book illness model then it is all in your head been-there-done-that. I was diagnosed about a year ago after several Oregon doctors turned their back on me not wanting to deal with the local medical political climate. The few who didn't turn me away hung in there with me and saved my life.
My new friend (from the ER waiting room) was diagnosed about 14 to 16 years ago in Eugene, Oregon. She was deathly ill, hospitalized and was first diagnosed with Guillain-Barre syndrome, yet they still did not know exactly what was going on with her. Her doctors were all puzzled.
She had one doctor who used her personal three days off from work researching the internet on her behalf and wondered if she might have porphyria. This doctor did the porphyria testing and sure enough she nailed the correct diagnosis of AIP. Those kind of doctors are few and far between.
What I find amazing about our chance encounter in the local ER waiting room was that we both found out we were afflicted with this very rare disease in a sparsely populated area. One of us being among only approximately 1 in 100,000 porphyria patients affected in the US. It makes our chance meeting even more amazing
My personal take on just how rare porphyria is: there are probably more porphyria patients out there that could be identified if our doctors had more exposure in their training about the disease; and lab technicians were trained to collect the specimens and process the samples correctly. That is my nickel's worth
If you have questions regarding testing, symptoms and treatment used for the Acute Porphyrias please visit: porphyriafoundation.org "Remember.Research is the key to your cure!"
Kathryn Nelson and PCT Journey!
Friday - April 22, 2016 @ 10:00:17
Kathryn Nelson
My Porphyria Cutanea Tarda (PCT) experience began roughly seven years ago. At the time I was living in Irving, Texas. When lesions appeared on my face, forearms and legs, I thought that perhaps it was related to Psoriasis, an autoimmune disorder I had since I was a child. As a result, I resorted to a fairly common approach to Psoriasis which is exposure to UV rays. I spent an hour or so every afternoon in the sun, but more lesions developed and the existing ones grew in size. Repeated visits to my Dermatologist resulted in a variety of diagnoses including eczema, hives and finally a "picking" disorder which basically meant the doctor believes you are picking at your skin causing sores and infections. His primary reason for this was that the sores appeared only where I could physically reach the area, in other words, there were no lesions on my back or the backs of my legs. I pointed out that as a forty plus year old woman, I didn't think I had suddenly started to pick my skin but he was not deterred.
I continued to use antibiotic ointments on the most severe patches and, as a result of other health concerns, discontinued my efforts to get a daily dose of UV rays. Over time, the lesions began to shrink but a new symptom developed. My skin darkened dramatically in areas where there were no blisters and where the blisters had healed, the skin turned white, giving the appearance of vitiligo. I was seeing a Rheumatologist for other autoimmune symptoms and he suggested I visit a dermatologist he knew who specialized in autoimmune skin disorders. He also prescribed Plaquenel to treat lupus-like symptoms tied to an undifferentiated connective tissue disorder.
It was several months later when I met with Dr. Melissa Costner in Dallas. By that time, most of the lesions and blisters were healed, however, the splotchy dark patches and scarred white areas covered most of my arms, face and large portions of my legs. I described my experiences over the previous three years. Dr. Costner listened closely and then said she wanted to do some blood work. Three weeks later, I returned to her office where she explained I had a familial type of PCT. In one minute, all of the heartaches of the previous four years made sense. She listened to me, something the previous doctors failed to do. She believed me when I said I wasn't "picking" my skin and said that rather than affecting areas where I "could reach," PCT was actually affecting areas being exposed to the sun. By reducing the time I was spending in the sun due to my energy issues, I had actually started the PCT healing process.
Today I still have very dark patches on my skin and very white scars but I have few reoccurrences of PCT. I play close attention to the medications I take, particularly since I have other auto-immune disorders which require a heavy regimen of treatment. PCT was not something I had ever heard of and was honestly not my biggest health concern at the time. But finding a doctor who listened gave me a sense of empowerment that I continue rely on today in all aspects of my health and with all my doctors.
"Remember.Research is the key to your cure!"
What Is AIP?
Thursday - April 21, 2016 @ 09:00:14
Acute Intermittent Porphyria (AIP)
This is one of the hereditary hepatic porphyrias. Its inheritance is autosomal dominant. The deficient enzyme is porphobilinogen deaminase (PBGD), also known as hydroxymethylbilane synthase (HMB synthase). This enzyme was formerly known as uroporphyrinogen I-synthase, and this term is still used by some clinical laboratories. A deficiency of PBGD is not sufficient by itself to produce AIP, and other activating factors must also be present. These include hormones, drugs and dietary changes. Sometimes, activating factors cannot be identified.
Most people who inherit the gene for AIP never develop symptoms. However, experts recommend that all relatives of someone with AIP obtain testing, to determine who has the genetic trait and who does not. Those who test positive for the trait should be educated as to measures that will help avoid attacks. Prevention is essential to good management.
AIP manifests after puberty, especially in women (due to hormonal influences). Symptoms usually come as discrete attacks that develop over two or more days. Abdominal pain, which is associated with nausea, can be severe and occurs in most cases.
Other symptoms may include:
nausea
vomiting
constipation
pain in the back, arms and legs
muscle weakness (due to effects on nerves supplying the muscles)
urinary retention
palpitation (due to a rapid heart rate and often accompanied by increased blood pressure)
confusion, hallucinations and seizures
Sometimes the level of salt (sodium and chloride) in the blood decreases markedly and contributes to some of these symptoms. The skin is not affected.
Diagnosis
Because this disease is rare and can mimic a host of other more common conditions, its presence is often not suspected. On the other hand, the diagnosis of AIP and other types of porphyria is sometimes made incorrectly in patients who do not have porphyria at all, particularly if laboratory tests are improperly done or misinterpreted. The finding of increased levels of delta-aminolevulinic acid (ALA) and porphobilinogen (PBG) in urine establishes that one of the acute porphyrias is present. If PBGD is deficient in normal red blood cells, the diagnosis of AIP is established. However, measuring PBGD in red blood cells should not be relied upon by itself to exclude AIP in a sick patient, because the enzyme is not deficient in red blood cells of all AIP patients.
If it is known that someone in a family has AIP, and their enzyme value is low in red blood cells, other family members who have inherited a deficiency of PBGD can be identified by measuring the enzyme in their red blood cells. Latent cases so identified can avoid agents known to cause attacks. However, in some AIP families, PBGD is normal in red blood cells and is deficient only in the liver and other tissues. Falsely low values sometimes occur due to problems with collecting and transporting the sample.
DNA is the material in cells that encodes all the genetic information of an individual. Many different mutations have been identified in the portion of DNA that comprises the gene for PBGD. However, within a given family, everyone has the same mutation. When that mutation is known for one member, screening of the relatives is straightforward and can be done on DNA from saliva (spit) or a swab of the inside of the cheek. This is now the gold standard of diagnosis and is available through specialty labs. Details are available from the APF or investigators of the national Porphyria Consortium http://rarediseasesnetwork.epi.usf.edu/porphyrias/index.htm.
Treatment and Prognosis
Hospitalization is often necessary for acute attacks, particularly if nausea and vomiting have prevented adequate oral intake. Medications for pain, nausea and vomiting, IV hydration, and close observation are generally required.
Glucose and other carbohydrates can help suppress disease activity, are given by vein or by mouth, and are part of initial treatment. Intravenous heme, however, is both more specific and effective than glucose and should be started if the patients symptoms fail to improve within 36 hours. Heme is sold as Panhematin, from Recordati Rare Diseases. Most hospitals do not stock it. Therefore the pharmacy must be notified at the time the patients admission to initiate a request for air-freighting enough medication for 5 days of treatment. Generally, shipping will take at least 24 hours.
Panhematin, is the only commercially available form of heme for treatment and prevention of acute porphyric attacks in the United States. Heme arginate, which is marketed in other countries as Normosang (Orphan Europe), is another preparation for intravenous administration. The main side-effect of Panhematin is irritation of the vein used for infusion (phlebitis). This is avoided by slow infusion through a large caliber vein or central line. Adding human albumin to the heme solution also may reduce the risk of phlebitis. (Directions for preparing Panhematin in this manner can be obtained from porphyria specialist and is included in the Primary Care Physician/Emergency Room Kit.) Heme therapy is indicated only if an acute attack of porphyria is proven by a marked increase in urine PBG. It may be useful also as preventive therapy for people with frequent recurrent attacks.
During treatment of an attack, attention should be given to salt and water balance. Harmful drugs should be stopped. These include barbiturates, sulfonamides, and many others (see the Acute Porphyria Drug Database). Attacks are often precipitated by low intake of carbohydrates and calories in an attempt to lose weight. Thus dietary counseling is very important (see below). Premenstrual attacks often resolve quickly with the onset of menses; hormone manipulations may prevent such attacks.
AIP is particularly dangerous if the diagnosis has not been made and if harmful drugs are administered. The prognosis is usually good if the disease is recognized and if treatment and preventive measures are begun before severe nerve damage has occurred. Although symptoms usually resolve after an attack, some patients develop chronic pain. Nerve damage and associated muscle weakness can improve over a period of months or longer after a severe attack. Mental symptoms may occur during attacks, but are usually not chronic.
Wearing a Medic Alert bracelet is advisable for patients who have had attacks. People who are asymptomatic carriers of the genetic trait may choose not to wear a bracelet but should be prepared in any medical encounter to advise their care-givers of medications that are risky in AIP. It should be remembered that AIP patients can develop other diseases, and symptoms will not always be due to porphyria.
Diet
AIP patients prone to attacks should eat a normal or high carbohydrate diet and should not greatly restrict their intakes of carbohydrate and calories, even for short periods of time. If weight loss is desired, it is advisable to consult a physician, who may request that a dietitian estimate an individual's normal caloric intake, which varies greatly from one person to another. It may be appropriate to prescribe a diet that is approximately 10% below the normal level of calories for the patient. This should result in a gradual weight loss and usually will not cause an attack of porphyria. (Please see the Diet and Nutrition section of our website for more information)
Having Children
Pregnancy is tolerated much better than was formerly believed. Offspring have a 50% chance of inheriting the gene for AIP, but the great majority of them remain "latent" for all or most of their lifetimes. The minority that eventually have symptoms usually benefit from treatment. Given these considerations, most patients or individuals with "latent" porphyria elect to have children for the same reasons as anyone else.
Special Interview: Justin Hamilton and his life with CEP~ Congenital Erythropoietic Porphyria
Wednesday - April 20, 2016 @ 16:32:46
Thank you Justin for participating in this interview. (Congenital Erythropoietic Porphyria) (CEP))
We take a close look at a day in the life of Justins Life with CEP, well also learn what it is and just how rare it really is. http://www.clinuvel.com/en/skin-science/skin-conditions/rare-skin-conditions/congenital-erythropoietic-porphyria-cep
So at what age did you start experiencing problems? What were they?
When I was first born. They noticed in the hospital my urine was a red fluorescent color. They thought it had something to do with my kidneys.
Did anyone else in your family have a history of CEP? No
When and how were you diagnosed? Where? When I was 6 months old. They did blood test and cultures. I was diagnosed at the McCook, Nebraska Clinic. (Currently 150 or less reported cases)
Did they get your diagnosis right the first time? Yes, besides thinking it was my kidneys. Mom then noticed blisters when the sun would be on me. They then did test to find out I had CEP.
How do you feel about having CEP the ups and downs? CEP sucks, I wouldn't wish it on my worst enemy if I had one. The ups to CEP there are not really any at all, other than that it gives you a look at life to strive to be positive, making you a fighter for the things that most people take for granted. The downs are endless and new every day. The fact that you are unable to go outside without being all covered up and drenched in sunscreen. The pain and the scarring that happens from any tiny bit of exposure. Not being able to be "normal" like others that can run around outside and soak up the sun. Growing up and living with CEP people staring at you and giving you mean looks because they are not aware of what really is going on. There are times that Im down and stressful times. Trying to cope with all that is thrown at you is a daily struggle.
What are your symptoms? How have you managed your symptoms? Symptoms are any exposure to the sun or certain lights I get blisters, some big some small. All very painful, and then after they pop they take weeks to heal. Then when they do heal they leave scars that limit normal skin growth. Pain becomes a normal thing that I become used too and just pray it helps fast each time. Trying to manage it is just lots of sunscreen and coverage when in the sun. I believe everything happens for a reason. So I just take what the good lord gave me and make the best of it.
Have you learned anything from having a rare disease?
CEP HSS taught me to take this disease seriously. I wish I would of been more protected when I was younger age and not been so stubborn when putting on sunscreen, long sleeved shirts, hats and so on. I seem to learn something new every day.
How have your Doctors treated you in the past and present?
They have treated me with antibiotics for infections from my sores. They have had me use steroid creams for my sores. Really that about it. This disease did attack one of my eyes. Which I now don't have much vision left in. Also caused a small hole in that eye. I see an eye specialist regularly. They put glue in the hole and a contact over to keep it stable. My doctors have been supportive and helpful with everything that comes with it.
What are your future goals? Hobbies that you enjoy?
My goals in life are to live as long as I can be watching my Kids growing up and spending time with my wife. I try to stay as covered as possible. And take each day like it was my last. I enjoy the outdoors which is hard since that is where I get most of my exposure from. But I live raising crops, animals, hunting, and fishing. My love for race cars keeps me going.
Do you work and what do you do?
In try to keep busy. Working hard is always been a big part of my family's way of life. I try my best to continue to help family and friends with what I am limited too. I try to help at the farm or the family car dealership when I can.
Are you Married & do you have children & how has this impacted you in any way?
I am happily married since 2011, since then we have two beautiful children. A boy Brayden who is 2 1/2 and a girl Brylee who is 1 1/2. They are my reason for living. I love then so much. There are some thing that limit me to my husband and father duties. With my hands being so small and sore (my hands are ½ the size of a normal persons due to the severe scaring) its hard for me to change diapers, simple stuff like that. I can't play with them outside on a nice day for a very long period of time like I would like to be able too.
If Holly would like to comment, how do you feel about Justin as a husband and father and how does he handle the CEP daily? He is an amazing husband and an even more amazing dad. There are some things that limit him. But we are aright with that. I understand and try to help him in every way possible to make things easier on him. He has good days and bad days. He does not shoe to anyone else how much pain he might be in. But when it is just the two of us he is able to open up some more. He is a very positive attitude about it all. Some days he may be in enough pain he stays in bed all day. But the next he is ready to go. The kids and I love him so much.
What encouragement would you give to others that have CEP and why raise awareness?
All I can say is words of encouragement. Don't take life for granted. Take your disease seriously and take the needed precautions no matter how much it sucks. Keep positive and have faith. There will be good and bad days but how you react to it is your choice. So stay positive and live life to the fullest. It is very important to spread awareness so that others may understand. The more people and doctors that know more about it they can understand to make it easier on me and ways to help.
If the APF has helped you at any given point, how have they helped you?
The APF has helped out greatly. The thing I like the most is it brings all fellow porphyrians together to tell their stories and help one another to make this disease better to live with. In am very thankful and blessed even with CEP.
Thank you Justin for sharing your life with CEP! Holly Thank you for raising awareness and your support to your husband and to the American Porphyria Foundation. For a description of CEP please read below also you can learn more information by contact the APF~ 1-866-APF-3635 or visit porphyriafoundation.org
Here are the facts of: Congenital erythropoietic porphyria, or CEP, is an extremely rare, inherited metabolic disorder. It is caused by genetic defects which lead to deficiency of the enzyme uroporphyrinogen III cosynthase (UROS). The disease is characterized by extreme photosensitivity (abnormal cutaneous reaction to sunlight) which can leave severe scarring, blister formation and the loss of digits or other features. Damaged skin can become infected, leading to further necrosis and deformities. The face, hands and arms are the most significantly affected as they are frequently exposed; sometimes presenting as severe disfiguration.
Incidence
The incidence of CEP is not known, but it is exceedingly rare; as of 2006, there were approximately 150 cases reported worldwide. Onset is usually during the early years of life as the genetic defects are present from birth. A milder form with onset in adulthood has been described in a small number of patients; six individuals as of 1990.
Causes
Congenital erythropoietic porphryia belongs to a family of diseases known as porphyrias. These are characterized by the overproduction and accumulation of chemicals, known as porphyrins, in various tissues within the body. The porphyrias come about due to faults or deficiencies in the pathway which leads to the production of heme (a major component of the hemoglobin in red blood cells).
CEP is caused by a genetic defect, or mutation, in the UROS gene. This gene encodes an enzyme, uroporphyrinogen III cosynthase, which is essential to the step-wise progression of heam synthesis (see Figure 1). The effect of the mutation is that the enzyme is only marginally active. This means that not only is heam synthesis severely impeded, but also that the pathway takes an alternative route, leading to the build of porphyrins. In the case of CEP, the particular porphyrins in excess are uroporphyrin I and coproporphyrin I.
Figure 1. The Heme Biosynthetic Pathway, illustrating the defect in CEP
While the enzymes activity is dramatically reduced in CEP patients, it is not entirely absent as this would likely result in the complete absence of red blood cells and therefore death. The porphyrins are partially excreted in the urine and feces, though a large portion enters into circulation and is deposited in bodily tissues; primarily the bone marrow, skin, blood and teeth. Porphyrin accumulation has toxic effects on the cells in these organs. This accounts for many of the symptoms experienced by CEP patients, including their heightened photosensitivity.
CEPs rarity is partially due to its pattern of inheritance as a recessive trait. This means that an individual needs to have two copies of the defective gene - one from their mother and one from their father - in order for the condition to be present. An individual with just one defective gene will be a carrier of the disease but will not exhibit any symptoms, since the properly functioning gene is able to produce intermediate levels of the enzyme. There is typically no family history of CEP and both parents are healthy.
A number of mutations in the UROS gene have been discovered in CEP patients, all cause dysfunction of the UROS enzyme. Of the 35 described, the C73R mutation is the most common, present in about a third of all cases. This particular mutation is associated with a severe form of the disease.
Symptoms
Symptoms of CEP are diverse and can range from mild to severe. Along with cutaneous damage due to enhanced photosensitivity, CEP patients commonly suffer anemia due to the breakdown of red blood cells (hemolysis). The following symptoms may be present to differing extents in individual patients:
Skin
·Blistering and rashes on light-exposed skin
·Scarring
·Increased skin fragility
·Skin destruction and erosion
·Abnormal hair growth (hypertrichosis/hirsutism)
·Loss of eyebrows and eyelashes
·Mutilation of cartilage structures, such as the ears and nose
·Loss of digits and facial features
·Bacterial infection of damaged skin, possibly leading to further necrosis and deformation
Blood and other tissues
·Anemia due to the breakdown of red blood cells
·Excessive red blood cell production (erythrocyte hyperplasia)
·Bone loss, fragility or hardening
·Enlarged spleen (splenomegaly)
·Brown, pink or red discoloration in urine, due to the presence of porphyrins
·Teeth stained red (erythrodontia), also due to accumulation of porphyrins
·Ocular (eye) lesions
Prenatal
·Brownish color to the amniotic fluid
·Accumulation of fluid in the fetus whilst stillin utero(hydrops fetalis)
In addition to the physical symptoms, CEP patients often suffer poor mental health. This is due to both the level of pain and discomfort experienced and the psychological impacts of their appearance. These individuals can be stigmatized and often avoid interaction with other people. Many struggle both socially and professionally, thus CEP has an extreme impact upon patient quality of life.
Diagnosis
CEP is the only porphyria that can be diagnosed prenatally. It is indicated by raised levels of uroporphyrin I in the amniotic fluid as early as 16 weeksin utero. More commonly, diagnosis is in infancy or childhood through a combination of the following methods:
·History of patient symptoms
·Quantitative screening using spectrophotometry or fluorimetry is considered the most accurate method of diagnosis. When uroporphyrin I and coporphyrin I are present in blood a plasma spectrofluorimetry is seen at 615-620 nm.
·Measurement of elevated levels of uroporphyrin I and coproporphyrin I in blood, urine or fecal analyses
·Examination of the eyes or urine using a Woods lamp
Treatment
Total avoidance of sunlight and other sources of visible/UV light (i.e. solariums) is vital to preventing skin damage in CEP. Sun-protective clothing and sunscreen containing the light-blocking compounds zinc oxide or titanium dioxide may offer some protection. Plastic window films and window tinting around the home and in the car can reduce damaging wavelengths of light penetrating the skin. In addition, incandescent light bulbs emit less phototoxic wavelengths of light than fluorescent lights. Physicians recommend CEP patients try to avoid physical injury which may worsen fragile or damaged skin.
CEP treatments can be radical and patient response is varied:
Bone marrow transplantation (BMT)- the only therapy to date that presents a potential cure for CEP. It has proved extremely successful in a handful of patients, however the long-term results of BMT are not yet known. This form of therapy also comes with significant risks, including infectious complications and a high mortality rate. In a few children, BMT has resulted in complete remission of CEP. Improvements included normal hemoglobin, considerably reduced uroporphyrin I in the urine and no skin lesions in spite of unrestricted sun exposure; this is evidence of the best case scenario. Stem cell cord blood transplantation has also been reported effective in a few CEP patients.
Blood transfusions -these decrease the production of red blood cells and hence porphyrins in CEP patients. Transfusions have successfully reduced disease symptoms in several patients, however there are complications associated with chronic transfusions (i.e. iron overload).
Splenectomy- a splenectomy is the surgical removal of the spleen. It can increase the lifespan of red blood cells, reducing anemia.
Oral sorbent medications- these include activated charcoal and cholestyramine which act by binding the excess porphyrins and preventing their absorption. In several patients these have reduced porphyrins in the blood and urine, in one case leading to complete remission. In contrast, there are also several reports of no effect and one of exacerbated condition; thus, caution should be exercised. Further, this treatment may be accompanied by the mild complication of poor nutrient absorption.
Oral beta-carotene- may act as a mild photo protectant to reduce the symptoms of photosensitivity, though treatment usually requires unreasonably large doses. It has been trialed in CEP with minimal efficacy.
Oxygen quenchers- these are oral medications, such as ascorbic acid and alpha-tocopherol, which mop up excess reactive oxygen species to lessen porphyrin-induced photo damage; also only marginally effective.
Prognosis
The prognosis for a CEP patient depends on both the clinical severity of the condition and their response to available treatment. Severity is mainly dependent on the amount of residual activity the UROS enzyme has. Into the future, potential therapies may exist in the form of stem cell transplantation and gene therapy to correct the mutation and produce a functional UROS enzyme.
During puberty, a childs hemoglobin levels increase to that of an adult. Along with an increase in heam comes a rise in porphyrin levels in CEP patients, therefore increase in symptoms or relapse may occur during adolescence. Where treatment is ineffective, the life expectancy of CEP patients is shortened.
·Hift, R.J, Meissner, P.N & Kirsch, R.E, 1993, The effect of oral activated charcoal on the course of congenital erythropoietic porphyria,The British Journal of Dermatology,129(1):14-17.
·Wiederholt, T, 2006, Identification of mutations in the uroporphyrinogen III cosynthase gene in German patients with congenital erythropoietic porphyria,Physiological Research,55(suppl. 2):S85-S92.
I wrote best for Porphyria week I thought you might like it. Porphyria: I suffer in silence most every day because The disease I have is so hard to explain! The frustration and fear that happens when The attack is here! The frustration comes from no one understanding the situation and the fear is there from the uneducated and the unaware, you honestly feel like no one really cares! Your blood pressure is high your muscles are weak your stomach is in so much pain you just can't sleep! You are your own advocate because this is so rare the doctors the hospitals just don't seem to care! This purple disease sometimes is so hard to bear! But I am strong and I continue along on my path for my highest and best! I learn as I go along sometimes I don't feel so strong but then I realize I have to be because this is so rare, you see! I will fight until the end and I hope with education care and love we will win! Sherrie Dawn "Remember.Research is the key to your cure!"
From Executive Director of the American Porphyria Foundation Desiree Lyon: Medical Moment: MEDICAL MOMENT~
Monday - April 18, 2016 @ 14:04:02
From Executive Director of the American Porphyria Foundation Desiree Lyon: Medical Moment: MEDICAL MOMENT~ I have had questions about Panhematin related to an article from 1970. First, the hematin today is not the same as forty years ago. Next , todays Panhematin was FDA approved in 1983 and has not had one death or viral report to the FDA in these many years. People can have side effects, nausea, headaches, flu feelings etc. But doctors continue to prescribe it and patients continue to take it because it stops attacks and the attack is far worse than the treatment. For those who have never had it , I am sure our friends here will tell you about it. It is given by infusion. I describe it by saying it looks like crank case oil. It is a greeny , blackish , swamp water looking semi fluid but it works like gold. I howled laughing recently when I read that some people accused me of trying to promote Panhematin because I was paid by the drug company. First, Panhematin is the only treatment we have to stop attacks. Glucose can help but when in a crisis, it is not suggested to stop an attack. so, of course I will tell ppl about it as exec dir of the apf. Next, no drug company pays me and to tell you the truth, it costs me to work for the APF. I went twenty plus years without a salaryâ?¦.so if these silly folks want to do my job, they will find no drug companies give you anything. the apf does take donations but if they come from industry, they are unconditional grants. If you have questions re Panhematin, you can pm me or ask the group. But be assured, an article from 1970 is not relative today if ever. Forward Ho !!!
read about Panhemtin here:
Panhematin is a treatment for the acute porphyrias manufactured by Recordati Rare Diseases in Lebanon New Jersey. It is a lyophilized form of alkaline heme that has to be reconstituted immediately prior to administration. Panhematin should be infused into a large peripheral vein. A large central line or port may be used, if available. Doctors administer Panhematin to correct heme deficiency in the liver and repress production of porphyrin precursors. Panhematin almost always normalizes porphyrin and porphyrin precursor values. Three to four mg/kg of Panhematin given once daily for four days early in an attack produces a highly beneficial effect in most patients. Commonly noted are decreases in pulse rate, blood pressure, abdominal pain, as well as decreased levels of urinary porphobilinogen (PBG). These effects can occur within a day. Panhematin is the only commercially available heme therapy in the United States. (Heme arginate is another preparation, but is only available outside of the U.S.) While a high car bohydrate diet is recommended for patients with porphyria, it is not regarded as highly effective by itself. Intravenous glucose therapy is a treatment option for mild attacks. When heme therapy was introduced as a treatment, it was recommended that it be initiated only after several days of glucose therapy was unsuccessful. Today, physicians experienced in treating patients with attacks of porphyria recommend early use of Panhematin rather than waiting to see if glucose alone will be of decisive help.
"Remember.Research is the key to your cure!"
APF Awareness Items for Sale:
Friday - April 15, 2016 @ 10:00:07
APF Awareness APF merchandise is going fast here is an updated is of sizes and items available: ALL ITEMS TO VIEW: https://www.facebook.com/amy.l.chapman.7 Long sleeve 25.00 ea BOGO 1/2 off SM-3 2xl-7 Med-3 3xl-8 Short Sleeve 19.00 ea BOGO 1/2 off S-11, Med-7, LG-10, XL-7, 2xl- 3, 3xl-2 Dont forget our NEW PURPLE APF Porphyria Strong wristbands 2/6 New Indoor/Outdoor APF Magnet 12.00 Small bottle APF Handmade necklace By Nona L 15.00 Large Bottle APF Handmade Necklace BY Nona L 20.00 Handmade APF key chain in Silver/Purple 10.00 All of these cover priority shipping! All proceeds go to APF 100% Get your's today- Show your APF Proud!
Email orders to Amy.APF@gmail.com VISA and MC accepted at this time
Show your APF- Porphyria Strong!
How Do 'Genetic Superheroes' Overcome Their Bad DNA?
Wednesday - April 13, 2016 @ 15:53:52
How Do 'Genetic Superheroes' Overcome Their Bad DNA?
A lucky few stay healthy despite carrying genetic defects linked to serious diseases. What protects them?
Leigh Wells/Getty Images/Ikon Images
Scientists say they've figured out how to track down people they call "genetic superheroes."
These are people who remain healthy even though they were born with genetic mutations that would usually lead to devastating disorders.
If enough of these people can be identified and studied, the researchers hope they could yield important new insights into the causes of many genetic disorders and possibly lead to new ways to prevent or treat them.
"This is a powerful opportunity to benefit many people by searching within one person to find something that could help many," says Dr. Stephen Friend, who runs Sage Bionetworks, a nonprofit research center in Seattle, and helped lead the work.
If it turns out that these people are carrying other genes that produce proteins that protect them, researchers might be able to use that information to develop useful new drugs, Friend and others say.
"This is an incredibly impressive paper," says Daniel MacArthur, who studies genetics at Harvard and MIT but wasn't involved in the work, which was published Monday inNature Biotechnology. He wrote a commentary accompanying the paper.
Researchers have long known that there are rare people who stay healthy despite profound genetic defects. Friend and his colleagues decided to systematically identify such individuals.
"We were interested in finding what might protect people from getting disease," Friend says. "If you're going to try to look at what protects people from getting disease, it is not necessarily the right thing to look at those who have disease. The right thing to do would be to look at those who have not gotten sick."
So Friend and colleagues at the Icahn School of Medicine at Mount Sinai in New York launched a massive search. They gathered genetic data from 586,306 people from around the world. The data came from private companies, hospitals, universities and elsewhere. The researchers then scanned the DNA sequences for known disease-causing mutations.
The researchers identified 13 people who had defects thought to always cause eight severe disorders, including cystic fibrosis. But none of the 13 individuals appeared to have developed any signs of these conditions, which Friend says is "very remarkable."
The absence of illness in these people suggests there may be something else in their genes, or in their environment, helping them. "There must be some protective element for them to have escaped the severity of the symptoms that would have been expected," Friend says.
Unfortunately, he says, none of the 13 had agreed ahead of time to allow researchers to contact them later for further studies. Ethical standards require prior consent to do that.
Had the subjects been asked to give consent upfront, researchers might have been able to determine what enabled them to escape what should have been their genetic fate.
"It's extremely frustrating not to be able to go back and to recontact them," Friend says.
But other researchers say the study is nevertheless important because it shows that advances in genetic and data analysis technologies have made it possible to find large numbers of these people. Because many genetic disorders are rare, large numbers of people would have to be found to yield useful information.
"If you'd asked me five years ago, it would have seemed laughable," MacArthur says. "But it seems entirely reasonable that over the next five years we will certainly have in the millions of individuals who could potentially be studied in this kind of work."
In fact, Friend and his colleagues have already identified a handful of other "genetic superheroes" through other means and have begun studying them. They include people who didn't develop Alzheimer's disease even though they are carrying mutations linked to the devastating brain disorder.
"We're actively decoding those individuals to identify how are they did they avoid getting that horrible disease," says Icahn's Eric Schadt, one of Friend's colleagues. "We're very excited about it."
The team is also also launching The Resilience Project, which aims to recruit millions of volunteers who have agreed in advance to let researchers study them.
"We hope to get tens of millions of people," Schadt says. "We think we have to be in the 1- to 10-million-person range to have a good shot to identify enough people across the different diseases to be effective at decoding and uncovering the reasons for their protection."
National Porphyria Awareness Week (NPAW) begins this Saturday, April 16th!
National Porphyria Awareness Week (NPAW) begins this Saturday, April 16th!
NPAW is the time of year that APF members spread porphyria awareness throughout their communities. It isn't too late for the APF to provide materials for your NPAW activity. We will also help you promote your event! ANY activity you partake in is important. It can be as simple as sharing a photo of you (or your beloved pet) wearing an APF t-shirt or headband!
The APF Pet Beauty Contest has been a huge hit and we want to make sure it ends with a bang! You have until Saturday, April 16th (the start of NPAW) to vote for your favorite pet. Votes are $1 each. You may view all the entries on the APF website. Below each entry is a direct link to their individual online voting page. Be sure to support your pet!
*Please contact the APF if you would like to host a meeting, too!*
"Remember.Research is the key to your cure!"
Order your APF supplies today for APF Awareness week!
Tuesday - April 5, 2016 @ 10:00:00
APF Awareness week is just around the corner!
This year~ We have new items and specials! If you would like to place an order please email your order to AMY.APF@gmail.com. I accept Visa/MC via email its secured and fast. Please include Size, Quantity, Color, Shirts- Long/Short Sleeve, Necklace, Color and size, and Key Chains.
From 4/10/16 to 4/23/16 FOR APF Awareness week:
White Short APF shirts- (only available in adults) sm-3xl-19.00ea
Long Sleeve APF shirts - (only available in adults) sm-3xl- 25.00ea
****Shirts are bogo 1/2 off at this time***** Less expensive shirt will be 1/2 off, first come first served!
APF Wristbands- Red, Blue, Purple, Dark Blue- 3.00 ea or 2/6$
Small Beach N -Bottle Necklace (Purple Sand, Shells, and hand made APF stamp) By Aritist Nona L. Bottle size is 1 inch tall- small 1 1/2 in tall for Large Necklace Length is 20 inches. small- 15.00 ea large is 20.00 ea Handmade Key Chain in Silver/Purple with APF listed is 10.00 ea
(since these are handmade please give 1-2 weeks for delivery
Car/Home Magnets- 3.8 x 8 Awareness Ribbon Shape Magnet Outdoor Safe 12.00 ea
All pictures are includes at the bottom. All prices included shipping Express Delivery USPS
All profits go directly to the APF! Be proud and show that your APF Strong!
RARE Champions of Hope Awards
Friday - April 1, 2016 @ 10:30:15
DO YOU KNOW A RARE CHAMPION OF HOPE WHO DESERVES PUBLIC RECOGNITION?
If so, submit your choice today!
Submit your choice of a RARE Champion of Hope here.
Global Genes 2016 Tribute to Champions of Hopes Community Nominated Awards are open for submissions today through April 30th! We are looking to YOU to nominate the individuals and organizations who are making a difference in the rare disease community.
· RARE Champion, Collaborations in Science & Technology
For more details on each award, please click here.
Nominations close on April 30th. Awards will be determined by nominees' merit, not by number of submissions.
The Tribute to Champions of Hopes Community Nominated Award recipients will be honored during a special evening event Friday, September 23rd as well as at the Annual Tribute to Champions of Hopes Gala, taking place on Saturday, September 24th. Both occasions are hosted at the Hyatt Regency Resort & Spa in Huntington Beach, CA.
Global Genes is a 501(c)(3) nonprofit organization working to eliminate the challenges of rare disease.
You Can Improve Your Health How?
Thursday - March 31, 2016 @ 13:46:04
You Can Improve Your Health
Rustam
RUSTAM, who lives in Russia, leads a busy life. In the past, he had some unhealthful habits but came to realize that he was paying a price for them. He stopped smoking and overindulging in alcohol. Still, long days in front of his computer left him feeling lethargic.
Although Rustam started work at eight oclock in the morning, he rarely felt fully awake until ten, and he was often sick. So he made an adjustment to his routine. The result? In the last seven years, I havent taken more than two sick days a year, he reports. I feel greatawake and alertand I enjoy life!
Ram, his wife, and their two small children live in Nepal. Sanitation is lacking in their neighborhood, and the area swarms with mosquitoes and flies. In the past, Ram and his family frequently suffered from respiratory problems as well as eye infections. They too made changes that greatly improved their health.
Take Control of Your Health!
Whether they are rich or poor, many people fail to see the link between their habits and their health. They may regard enjoying good health as a matter of chance or as something over which they have little control. Such a fatalistic view holds many back from improving their health and leading a more productive life.
In reality, whatever your financial circumstances, there are basic steps you can take to protect and greatly improve your own health and that of your family. Is doing so worth the effort? By all means! You can increase the quality of your life and avoid needlessly shortening it.
By word and example, parents can teach their children to form good habits, resulting in better health. The extra time and expense involved will be repaid in reduced suffering, less time lost to illness, and less money spent on medical bills. As the saying goes, An ounce of prevention is worth a pound of cure.
In the following articles, we will consider five basic keys that have helped Rustam, Ram, and many others. These keys can help you too!
"Remember.Research is the key to your cure!"
Desiree Lyon Howe to receive the NORD Rare Impact Award
Wednesday - March 30, 2016 @ 10:30:16
Desiree Lyon Howe to receive the NORD Rare Impact Award
Congratulations!!
Desiree Lyon Howe will be receiving the Rare Impact Award in Washington, DC on May 17, 2016 for her many accomplishments to further porphyria research, awareness and education. This prestigious award is given on behalf of the National Organization for Rare Diseases and the 30 million Americans they represent.
Desiree's response to the news was, "The staff and the members of the American Porphyria Foundation work together to make these advances possible. I share this Award with each of them."
WHEN TRAGEDY STRIKESHOW YOU CAN COPE Loss Of Health A personal Example!
Monday - March 28, 2016 @ 10:00:27
COVER SUBJECT WHEN TRAGEDY STRIKESHOW YOU CAN COPE
Loss Of Health
Mabel, in Argentina, led an active life and worked as a physical-rehabilitation therapist. In 2007, she began to feel especially tired and to have severe headaches daily. I went to several doctors and tried all kinds of medications, she says, but nothing helped. Finally, Mabel had an MRI scan, which showed that she had a brain tumor. She says: I was stunned! I couldnt believe that I had been living with this enemy inside me.
Still, I didnt really understand how serious my situation was until after I had surgery. When I woke up in intensive care, I couldnt move. All I could do was stare at the ceiling. Before the surgery I had been active and independent. Suddenly I could do nothing. My days in intensive care were filled with confusion and noise from medical equipment, emergency alarms, and moans from other patients. I felt as though I could breathe the pain and suffering in the air.
Today, I have recuperated to a certain degree. I can walk without help and even go out by myself at times. But I have double vision and still lack muscle coordination.
COPING WITH TRAGEDY
Maintain a positive viewpoint. At Proverbs 17:22, the Bible says: A joyful heart is good medicine, but a crushed spirit saps ones strength. Mabel recalls: During my recovery, I faced the same challenges that my own patients had faced. The exercises were very painful, and at times I thought of giving up. I had to force myself to dismiss such negative thinking, knowing that the effort would eventually bring good results.
Build hope in order to endure. From the Bible, I knew why tragedies occur, says Mabel. But I also knew that with each passing day, we are closer to the time when pain will be gone forever.*
Mabel recalls how this helped her: When they took me in for surgery, I experienced the truth ~ Do not be afraid, for I am with you. I felt an immense peace knowing that God cared about what happened to me.
"Remember.Research is the key to your cure!"
Acute Porphyria Emergency Reminders
Thursday - March 24, 2016 @ 10:30:08
THE PORPHYRIA RESEARCH CONSORTIUM of the Rare Disease Clinical Research Network of the NIH As a reminder, the Porphyria Research Consortium is a team of experts who are conducting the major life saving research for all of us. Dr. Montgomery Bissell, University of California, San Francisco; Dr. Karl Anderson, University of Texas Medical Branch; Dr. John Phillips,University of Utah; Dr. Herbert Bonkovsky, University of North Carolina; Dr. Joseph Bloomer, University of Alabama and Dr. Robert Desnick, Mount Sinai School of Medicine. Desnick Bloomer Anderson Phillips Bonkovsky Bissell Research volunteers are needed for all types of porphyria, including a research project for a new Alnylam treatment for the acute porphyrias. It only takes one day out of your life, please call the APF office asap and become a Medical Hero. YOU are the KEY because RESEARCH IS THE KEY TO YOUR CURE. Call the APF 1.866 APF.3635 EMERGENCY ROOM GUIDELINES KEY POINTS The APF website has an Emergency Room Guidelines section on acute porphyrias designed for primary care and emergency room physicians. The following Key Points are essential for diagnosis and treatment: 1. The human porphyrias are clinical disorders reflecting defects in heme biosynthesis. 2. Acute porphyrias cause acute attacks of neurological symptoms that can be life-threatening. 3. Acute attacks are triggered by certain drugs, sex steroid hormones, reduced intake of calories and carbohydrate, alcohol and unknown factors. 4. Many of these factors stimulate heme synthesis in the liver, which in the face of a metabolic enzyme defect, leads to increased production of heme precursors that may be neurotoxic. 5. Delta-aminolevulinic acid (ALA) and porphobilinogen (PBG), are porphyrin precursors and intermediates in the heme biosynthetic pathway. 6. ALA and porphobilinogen (PBG) are almost always elevated in urine during an acute attack of porphyria. 7. The most common emergency room (ER) clinical presentation is acute abdominal pain. Other features may include seizures, confusion and hallucinations, and a progressive polyaxonal motor neuropathy, which can progress to paralysis and respiratory failure requiring a ventilator. 8. A high index of suspicion in the presence of nonspecific symptoms is important for diagnosis. A family history of porphyria, female sex, onset during the luteal phase of the menstrual cycle, or recent use of a porphyrinogenic drug may be diagnostic clues. 9. A new diagnosis of porphyria as the cause of acute symptoms must be substantiated by finding a substantial increase in urine porphobilinogen (PBG). 10. Treatment should start promptly after the diagnosis is made. Mild attacks are sometimes treated with glucose loading (e.g. 3L of 10% glucose daily by vein). 11. Most acute attacks should be treated with hemin (Panhematin), Recordati Rare Diseaes at: www.recordatirarediseases.com or 866-654-0539; 3-4mg/kg into a large peripheral vein or venous access port daily for 4 days. Reconstituting Panhematin with human serum albumin rather than sterile water is recommended prior to infusion. This helps prevent phlebitis at the site of intravenous infusion. 12. Hospitalization is usually required for symptomatic treatment of pain, nausea and vomiting. "Remember.Research is the key to your cure!"
Are you ready for NPAW 2016? We are ready to raise Awareness!
Tuesday - March 22, 2016 @ 10:30:03
National Porphyria Awareness Week 2016
This is the time of year that each of YOU has the opportunity to reach out to advance porphyria awarenss in your local and medical communities. YOU are key to spreading knowledge about porphyria. For example, the Cook family has enhanced awareness in Vernon, TX by hosting a Barrel Race and educating kids in school about EPP. Amanda Boston teaches her nurses and doctors about porphyria with her every hospitalization. Claire and Bob Sadowniczak manned the APF porphyria booth at the Hematology Convention. Louise Schlosser is hosting a patient support meeting. Amy Chapman writes a blog and Rob Saupe' assists with the APF website, Amy Rose Burke is a Facebook administrator. There are countless ways to enhance awareness. The APF will provide you with materials to help with your venue and will be glad to make suggestions to accomplish your goals if you need assistance. There are many ways to spread awareness in your own community, including the suggestions below:
Tell your story to the local television, radio or newspaper
Share your knowledge with doctors or at your hospital seminars
Set up exhibit booths at local health fairs or medical meetings
Assist the APF at national medical conventions
Volunteer your talents to help achieve the APF patient educational projects
Host a community race or car wash to benefit APF physician education
Volunteer your skills, like computer skills, or business acumen
Host a Fun Run or Walk-A-Thon or even a Night Run or Twilight Walk
Share your story in the Member Story Section of the APF website
What are you going to do this year to raise awareness?
Go Local~ IF you need ideas, help or supplies from the APF please give us a call NOW were only a few short weeks away 1-866-apf-3635
"Remember.Research is the key to your cure!"
NIH Overview of ALL Porphyrias
Tuesday - March 15, 2016 @ 11:18:11
What is porphyria?
Porphyria is a group of disorders caused by abnormalities in the chemical steps that lead to heme production. Heme is a vital molecule for all of the body's organs, although it is most abundant in the blood, bone marrow, and liver. Heme is a component of several iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood).
Researchers have identified several types of porphyria, which are distinguished by their genetic cause and their signs and symptoms. Some types of porphyria, called cutaneous porphyrias, primarily affect the skin. Areas of skin exposed to the sun become fragile and blistered, which can lead to infection, scarring, changes in skin coloring (pigmentation), and increased hair growth. Cutaneous porphyrias include congenital erythropoietic porphyria, erythropoietic protoporphyria, hepatoerythropoietic porphyria, and porphyria cutanea tarda.
Other types of porphyria, called acute porphyrias, primarily affect the nervous system. These disorders are described as "acute" because their signs and symptoms appear quickly and usually last a short time. Episodes of acute porphyria can cause abdominal pain, vomiting, constipation, and diarrhea. During an episode, a person may also experience muscle weakness, seizures, fever, and mental changes such as anxiety and hallucinations. These signs and symptoms can be life-threatening, especially if the muscles that control breathing become paralyzed. Acute porphyrias include acute intermittent porphyria and ALAD deficiency porphyria. Two other forms of porphyria, hereditary coproporphyria and variegate porphyria, can have both acute and cutaneous symptoms.
The porphyrias can also be split into erythropoietic and hepatic types, depending on where damaging compounds called porphyrins and porphyrin precursors first build up in the body. In erythropoietic porphyrias, these compounds originate in the bone marrow. Erythropoietic porphyrias include erythropoietic protoporphyria and congenital erythropoietic porphyria. Health problems associated with erythropoietic porphyrias include a low number of red blood cells (anemia) and enlargement of the spleen (splenomegaly). The other types of porphyrias are considered hepatic porphyrias. In these disorders, porphyrins and porphyrin precursors originate primarily in the liver, leading to abnormal liver function and an increased risk of developing liver cancer.
Environmental factors can strongly influence the occurrence and severity of signs and symptoms of porphyria. Alcohol, smoking, certain drugs, hormones, other illnesses, stress, and dieting or periods without food (fasting) can all trigger the signs and symptoms of some forms of the disorder. Additionally, exposure to sunlight worsens the skin damage in people with cutaneous porphyrias.
How common is porphyria?
The exact prevalence of porphyria is unknown, but it probably ranges from 1 in 500 to 1 in 50,000 people worldwide. Overall, porphyria cutanea tarda is the most common type of porphyria. For some forms of porphyria, the prevalence is unknown because many people with a genetic mutation associated with the disease never experience signs or symptoms.
Acute intermittent porphyria is the most common form of acute porphyria in most countries. It may occur more frequently in northern European countries, such as Sweden, and in the United Kingdom. Another form of the disorder, hereditary coproporphyria, has been reported mostly in Europe and North America. Variegate porphyria is most common in the Afrikaner population of South Africa; about 3 in 1,000 people in this population have the genetic change that causes this form of the disorder.
What genes are related to porphyria?
Each form of porphyria results from mutations in one of these genes: ALAD, ALAS2, CPOX, FECH,HMBS, PPOX, UROD, or UROS.
The genes related to porphyria provide instructions for making the enzymes needed to produce heme. Mutations in most of these genes reduce enzyme activity, which limits the amount of heme the body can produce. As a result, compounds called porphyrins and porphyrin precursors, which are formed during the process of heme production, can build up abnormally in the liver and other organs. When these substances accumulate in the skin and interact with sunlight, they cause the cutaneous forms of porphyria. The acute forms of the disease occur when porphyrins and porphyrin precursors build up in and damage the nervous system.
One type of porphyria, porphyria cutanea tarda, results from both genetic and nongenetic factors. About 20 percent of cases are related to mutations in the UROD gene. The remaining cases are not associated with UROD gene mutations and are classified as sporadic. Many factors contribute to the development of porphyria cutanea tarda. These include an increased amount of iron in the liver, alcohol consumption, smoking, hepatitis C or HIV infection, or certain hormones. Mutations in theHFE gene (which cause an iron overload disorder called hemochromatosis) are also associated with porphyria cutanea tarda. Other, as-yet-unidentified genetic factors may also play a role in this form of porphyria.
Some types of porphyria are inherited in an autosomal dominant pattern, which means one copy of the gene in each cell is mutated. This single mutation is sufficient to reduce the activity of an enzyme needed for heme production, which increases the risk of developing signs and symptoms of porphyria. Autosomal dominant porphyrias include acute intermittent porphyria, most cases of erythropoietic protoporphyria, hereditary coproporphyria, and variegate porphyria. Although the gene mutations associated with some cases of porphyria cutanea tarda also have an autosomal dominant inheritance pattern, most people with this form of porphyria do not have an inherited gene mutation.
Other porphyrias are inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but do not show signs and symptoms of the condition. Porphyrias with an autosomal recessive pattern of inheritance include ALAD deficiency porphyria, congenital erythropoietic porphyria, and some cases of erythropoietic protoporphyria.
When erythropoietic protoporphyria is caused by mutations in the ALAS2 gene, it has an X-linked dominant pattern of inheritance. The ALAS2 gene is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the gene in each cell may be sufficient to cause the disorder. In males (who have only one X chromosome), a mutation in the only copy of the gene in each cell causes the disorder. Males may experience more severe symptoms of the disorder than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Mutations in the UROD gene are related to both porphyria cutanea tarda and hepatoerythropoietic porphyria. Individuals who inherit one altered copy of the UROD gene are at increased risk for porphyria cutanea tarda. (Multiple genetic and nongenetic factors contribute to this condition.) People who inherit two altered copies of the UROD gene in each cell develop hepatoerythropoietic porphyria.
Where can I find information about diagnosis or management of porphyria?
These resources address the diagnosis or management of porphyria and may include treatment providers.
The resources on this site should not be used as a substitute for professional medical care or advice. Users seeking information about a personal genetic disease, syndrome, or condition should consult with a qualified healthcare professional. See How can I find a genetics professional in my area? in the Handbook.
Cutaneous porphyrias primarily affect the skin. Areas of skin exposed to the sun become fragile and blistered, which can lead to infection, scarring, changes in skin coloring (pigmentation), and increased hair growth. Cutaneous porphyrias include congenital erythropoietic porphyria, erythropoietic protoporphyria and X-linked protoporphyria, porphyria cutanea tarda, and hepatoerythropoietic porphyria.
PCT is a deficiency of the enzyme uroporphyrinogen decarboxylase. Cutaneous blisters develop on sun-exposed areas of the skin, such as the hands and face. The skin in these areas may blister or peel after minor trauma. Increased hair growth, as well as darkening and thickening of the skin may also occur. Neurological and abdominal symptoms are not characteristic of PCT.
Liver function abnormalities are common, but are usually mild. PCT is often associated with hepatitis C infection, which also can cause these liver complications. However, liver tests are generally abnormal even in PCT patients without hepatitis C infection. Progression to cirrhosis and even liver cancer occurs in some patients.
Who gets PCT?
Porphyria cutanea tarda (PCT) is the most common type of porphyria, with a prevalence of approximately 1 case for every 10,000 people. PCT develops when the activity of an enzyme involved in synthesis of heme, uroporphyrinogen decarboxylase (URO-decarboxylase), becomes severely deficient (less than 20% of normal activity) in the liver. In most cases of PCT, patients do not have inherited URO-decarboxylase gene mutations and are said to have sporadic (or Type I) PCT (s-PCT) A URO-decarboxylase inhibitor generated only in the liver accounts for the severely deficient enzyme activity in s-PCT. About 20 percent of cases have familial (or Type II) PCT (f -PCT). Such individuals have inherited a URO-decarboxylase gene mutation from one parent, which has reduced the amount of URO-decarboxylase in all tissues from birth. However, to develop PCT symptoms, other factors must be present to further reduce URO-decarboxylase level in the liver to less than 20% of normal. Such f-PCT patients may develop blisters at an early age or have relatives with the disease. Excess iron and multiple other susceptibility factors contribute to the development of PCT. These susceptibility factors include excess use of alcohol, use of estrogens, chronic hepatitis C, smoking, HIV (human immunodeficiency virus) infections, and mutations of the HFE gene which is associated with the disease hemochromatosis in which excess iron accumulates in the liver. Other susceptibility factors exist but are as of yet unidentified.
What causes PCT?
PCT develops when the activity of the enzyme uroporphyrinogen decarboxylase (URO-decarboxylase) becomes severely deficient (less than 20% of normal) in the liver.
How is PCT Diagnosed?
The diagnosis of PCT can be made by finding an abnormally high concentration of porphyrins in urine or plasma. Abnormally high porphyrins in feces may also be observed.
Patients with f-PCT usually have no family history of the disease, but can be recognized by finding half-normal URO-decarboxylase activity in red blood cells, or preferably by DNA studies.
PCT, both familial and sporadic, can be treated either with regularly scheduled phlebotomies (removal of blood) to lower the amount of porphyrins in the liver or with a low dose regimen of hydroxychloroquine. In addition, the susceptibility mentioned above (see Who gets PCT?) need to be eliminated. PCT is the most treatable of the porphyrias. Treatment seems to be equally effective in f-PCT and s-PCT. Relapses that occur after the initial treatment can be treated in the same manner as the initial treatment.
"Remember.Research is the key to your cure!"
Erythropoietic Protoporphyria (EPP) and X-Linked Protoporphyria (XLP)
Wednesday - March 9, 2016 @ 10:30:16
Erythropoietic Protoporphyria (EPP) and X-Linked Protoporphyria (XLP)
Erythropoietic Protoporphyria is a disease of porphyrin metabolism characterized by abnormally elevated levels of protoporphyrin IX in erythocytes (mature blood cells), feces and plasma (the fluid portion of circulating blood), and by sensitivity to visible light.
This sensitivity manifests itself as a burning sensation in the skin, followed by varying degrees of erythema (redness of the skin due to capillary dilation) and edema (swelling caused by excess fluids). Consequently, patients with the disorder commonly avoid exposure of the skin to strong light. They tend to choose indoor occupations and nocturnal work, or to venture outdoors while heavily clothed to protect the skin.
Diagnosis
EPP is diagnosed in patients with light sensitivity by testing blood and stool for the presence of abnormally high levels of protoporphyrin. Contrary to what is found in the other porphyrias, urine porphyrin levels remain within normal limits in EPP. When a smear of blood from a patient is examined under the fluorescence microscope, large numbers of red fluorescing erythrocytes are seen; these are not seen in persons who do not have this disorder. In addition, if the skin of the light-exposed areas of the body is examined under the light microscope, an amorphous homogeneous substance in and around the walls of small blood vessels of the upper papillary dermis will be seen. Histologic studies suggest that this substance is a neutral mucopolysaccharide, glycoprotein or mucoprotein. EPP is genetically transmitted as an autosomal recessive trait. Some relatives of patients may also have only slightly elevated levels of protoporphyrin but are asymptomatic, suggesting the existence of a carrier state.
Clinical Features
Most EPP patients experience the onset of photosensitivity before the age of six years and some as early as eighteen months. Patients report, in decreasing order of frequency, burning, swelling, itching and redness of the skin. After severe episodes of photosensitivity, some patients acquire shallow-depressed scars over the nose and cheeks and on the backs of hands. Some patients report only subjective symptoms of itching and burning and have no redness, swelling or scarring; these patients are often dismissed by their physicians as hypochondriacs, when in reality they have EPP. Thus, it is important for the physician to investigate for the presence of the disease in all patients who report itching and burning of the skin on exposure to light, even in the absence of objective findings. The amount of exposure to sun that a patient with EPP can tolerate varies from a few minutes to several hours. This photosensitivity is to light in the visible spectrum (400 to 700 nm). These wavelengths are not absorbed by window glass. Therefore, the symptoms can also develop from light passing through glass windows. About half of the patients report decreases in photosensitivity during winter. However, those engaging in skiing report that the light reflected by snow causes severe photosensitivity reactions. EPP is generally a benign disease. Many patients have somewhat decreased levels of hemoglobin and hematocrit (percentage of the volume of a blood sample occupied by cells). This finding usually requires no treatment. One reported case with severe hemolytic anemia (anemia caused by excessive destruction of red blood cells) improved after splenectomy (removal of the spleen). There also seems to be an increased frequency of cholelithiasis (presence of the formation of solid material in the gall bladder or bile duct), with several patients requiring cholecystectomy (removal of the gall bladder). Chemical analysis of the gallstones reveals high levels of protoporphyrin. To summarize, in EPP a decreased amount of the enzyme ferrochelatase leads to the accumulation of protoporphyrin in reticulocytes (young red blood cells that appear especially during regeneration of lost blood). This excess protoporphyrin leaks rapidly into the plasma from the maturing reticulocytes and young erythrocytes. The protoporphyrin is then partially cleared from the plasma by the liver and excreted into the bile (with or without some recirculation via the enterohepatic circulation). Accumulation of this protoporphyrin in the liver may lead, in rare cases, to serious liver disease.
Treatment
Patients with EPP have found that topical sunscreens, which are effective in protecting against hypersensitivity to the sunburn spectrum of light, are ineffective as protective agents. Various systemic agents, such as antimalarials, inosine and vitamin E, have been tried but with little success. Orally administered pharmaceutical grade beta-carotene has been found to improve the photosensitivity associated with the disease. The majority of patients are able to increase their ability to tolerate sunlight by at least three times after taking beta carotene. (LUMITENE, Tishcon Corp., 60 to 180 mg per day, by mouth.) No side effects have been reported other than transient loose stools in a few patients and carotenodermia (yellowing of the skin), which was not cosmetically offensive to the majority of the patients. To order [url=/testing-and-treatment/medications-for-porphyria/lumitene]LUMITENE[/url] by email, contact [url=mailto:info@epic4health.com]info@epic4health.com[/url]. It is important for EPP patients to ingest the proper formulation of beta-carotene to obtain its greatest beneficial effect. It is also important to make sure that the preparation is a pharmaceutical grade formulation. The form having the highest effective absorbtion is the "dry beta carotene beadlets, 10" manufactured by Hoffmann-LaRoche. These are used in LUMITENE, the Tishcon Corporation's preparation previously discussed. You may order LUMITENE directly from E.P.I.C., a subsidiary of the Tishcon Corporation. Their telephone number for EPP patients calling from within North America is 1-800-866-0978. Vitamin A toxicity will never occur from the ingestation of high doses of beta-carotene. Please also note that preparations using beta-carotene crystals dissolved in vegetable oil are not suitable for use in treating EPP because these preparations are erratically absorbed by the body. A high intake of carotenoid-containing foods as a method of obtaining high levels of blood and skin carotenoids is not recommended. Toxic reactions, such as leukopenia (any situation where the number of leukocytes in the circulating blood is less than normal) and methemoglobinemia (presence of metheglobin in the blood) occur in those who ingest large quantities of vegetables in the amounts that would be necessary to obtain a protective effect. When purified beta-carotene is used, neither of these toxic reactions has occurred, indicating that the reactions were probably due to the non-carotenoid constituents of the vegetables. Patients with EPP may develop liver abnormalities due to an excess deposition of protoporphyrin in that organ. Hence, drugs that can impair bile flow, cholestasis, as well as estrogens, should be given cautiously. Cholestyramine ingestion may lower porphyrin levels in some patients. Some patients with EPP report that drinking alcoholic beverages increases their photosensitivity. It is probably wise for all EPP patients to avoid or greatly reduce alcohol consumption. Individuals with EPP may also need to wear protective clothing, such as garments with long sleeves and trousers: each one has to decide the amount of this sort of protection needed. Certain kinds of fabric, such as denim, are quite light protective. Also certain plastic materials, such as Scotchtint, filters out harmful rays and can be used on home and automobile windows.
2016 Pet Beauty Contest
Monday - March 7, 2016 @ 10:30:37
2016 PET BEAUTY CONTEST
Back by popular demand - we are hosting the 2nd Pet Beauty Contest. We can have FUN and FUNdraise to enhance Porphyria Awareness.
To enter the contest, all you need to do is mail or email your pet's name and photograph to the APF. Pigs, dogs, cats, hamsters, snakes, goats, cows, etc. - all pets are welcome, because all pets are beautiful!!! It is FREE to enter the competition! Your pet's photo will be posted on the APF website for all to see.
Once your pet's photo is on the website, ask friends to vote for your pet with a $1 donation per vote and return the votes to the APF by April 16th, the beginning of National Porphyria Awareness Week. You can collect your pet's votes individually or together and send them to the APF.
There are two categories of winners:
1. The pet who collects the most votes/donations;
2. The pet who gets the most people to vote/donate.
The winners will receive wonderful trophies and a story of their pet in the next newsletter and APF media sites. Let the FUN begin!!!!
SEND YOUR PICTURES AND VOTES TO:
porphyrus@porphyriafoundation.com
4900 Woodway Dr, Suite 780, Houston, TX 77056
Visit the website for contest updates! Make sure to scroll down to see all of the entries as they are added!
*Other APF members have scheduled patient meetings around the country for National Porphyria Awareness Week (April 16-23). Please let us know if you'd like to host a patient meeting in your area. More information on these meetings will be coming soon!
"Remember.Research is the key to your cure!"
Health of the mind Poem
Thursday - March 3, 2016 @ 10:30:04
Updated Porphyria Overview please share
Monday - February 29, 2016 @ 17:23:34
Porphyria is the umbrella term for a group of rare disorders that involve a particular molecule called heme or haem. Heme contains iron and is used in metabolic processes throughout the body. Porphyria occurs when the body cannot convert naturally occurring compounds (called porphyrins) into heme.
While all tissues have heme, those that use it the most are the red blood cells, liver and bone marrow. Porphyria can affect the skin, nervous system and gastrointestinal system, depending on the specific type.
In most cases, the cause is a combination of genetic and environmental factors. More women than men are affected for reasons unknown. There is no cure but treatments are available to manage the symptoms.
Symptoms
Symptoms vary from one type of porphyria to the next. Cases are generally classified into one of three groups, which include:
Acute porphyrias the condition mostly affects the nervous system. The skin is occasionally affected. Symptoms may include muscle pain or paralysis, seizures, disorientation, hallucination, bloody (red) urine, hypertension and gastrointestinal problems such as vomiting, abdominal pain and constipation. Acute porphyrias generally occur during adulthood and are rare before puberty or after menopause. Different types of acute porphyria include acute intermittent porphyria and erythropoietic protoporphyria.
Cutaneous porphyrias the condition affects the skin but not the nervous system. The skin is highly sensitive to sunlight and exposure tends to trigger symptoms within minutes. Symptoms may include red, itchy, blistered, painful and swollen skin and bloody (red) urine. The condition may develop during childhood. Different types of cutaneous porphyria include porphyria cutanea tarda and hepatoerythropoietic porphyria.
Neurocutaneous porphyrias the condition affects both the skin and the nervous system. Sunlight exposure tends to rapidly trigger symptoms. Different types of neurocutaneous porphyria include variegate porphyria and hereditary coproporphyria.
"Remember.Research is the key to your cure!"
Porphyrins build up in the body
The substance heme (or haem) is used in various metabolic processes. The body makes heme from porphyrins, which are metallic compounds found naturally in the tissues of animals and plants. The conversion of porphyrins into heme requires the action of special proteins called enzymes. Genes control the action of enzymes. A flawed gene (or genes) can stop the body from making one or more of these enzymes. This creates a lack of heme and a build-up of porphyrins, which causes the signs and symptoms of porphyria.
Inherited genes
Most forms of porphyria are inherited, which means the genetic predisposition is passed from one generation to the next. The faulty gene interferes with the bodys ability to create one or more enzymes necessary in the conversion of porphyrins into heme. The pattern of inheritance may include:
Autosomal dominant inheritance the faulty gene is inherited from one parent. This faulty gene overrides the healthy gene inherited from the other parent.
Autosomal recessive inheritance the faulty gene is inherited from both parents.
However, about nine in every 10 people with the faulty gene or genes dont have porphyria. It appears that an environmental trigger is needed to allow porphyria to develop.
A range of triggers
Various triggers can prompt the development of porphyria. While the factors in the following list may seem to have nothing in common, each one demands increased heme production, which overwhelms the bodys ability to deal with the increased levels of porphyrins.
Common triggers include:
Prescription drugs such as barbiturates, tranquillisers, sedatives, oral contraceptives and some types of antibiotics
Female sex hormones
Sunlight
Alcohol
Cigarette smoking
Infection
Surgery
Fasting.
Common complications
Without medical treatment, complications of porphyria may include:
Permanent hair loss
Skin scarring
Permanent skin pigmentation changes
Dehydration
Breathing problems
High blood pressure (hypertension)
Low salt levels in the blood (hyponatremia)
Kidney failure
Liver problems, which may require a liver transplant in severe cases.
Diagnosis
Since porphyria is rare, most doctors are unfamiliar with it and may not recognize the symptoms. Diagnosis can be delayed because porphyria mimics the symptoms and signs of various other medical conditions such as Guillain-Barre syndrome, eczema, multiple sclerosis and irritable bowel syndrome. Diagnostic tests may include:
Physical examination
Medical history
Urine tests to check for elevated substances including porphyrins
Blood tests to check for high levels of porphyrins in the plasma
Stool sample to check for excreted porphyrins
Genetic test.
Treatment acute porphyria
Treatment may include:
Pain medication
Addressing the underlying cause for example, prescribing antibiotics to treat an infection or ceasing a particular medication
Medication called hematin, which is a type of heme the body can use
Intravenous fluids and glucose
Admission to hospital in severe cases.
Treatment cutaneous porphyria
Treatment may include:
Oral administration of activated charcoal, which helps to absorb excess porphyrins
Daily supplementation with beta-carotene (vitamin A) as part of long-term treatment.
Self-care options
Be guided by your doctor, but general suggestions include:
In all cases avoid known triggers for example, dont smoke.
When out in the sun, wear sunglasses, a brimmed hat, a long-sleeved top and long pants. Apply SPF 30+ sunscreen to exposed skin areas.
Protect your skin every day. For example, wear rubber gloves when handling chemicals or very hot water. Avoid perfumed soaps. Regularly apply barrier cream to the hands.
Eat regular meals and avoid alcohol.
You may like to consider wearing a medical alert bracelet or pendant, since surgery and some drugs can provoke symptoms.
Where to get help
Your doctor
In an emergency, call triple zero (000)
NURSE-ON-CALL Tel. 1300 60 60 24 for expert health information and advice (24 hours, 7 days)
Porphyria Association Inc. Tel. (03) 9845 2737
Genetic Support Network of Victoria Tel. (03) 8341 6315
Australasian Genetic Alliance Tel. (02) 9211 1462
Things to remember
Porphyria is the umbrella term for a group of rare disorders that involve a particular molecule called heme or haem.
Porphyria can affect the skin, nervous system, gastrointestinal system or all of these, depending on the specific type.
There is no cure, but medical treatment and lifestyle changes can usually manage the symptoms.
RARE DISEASE DAY IS TODAY
Monday - February 29, 2016 @ 10:30:03
No matter where you are in this world, please share your support and love for all of those who are RARE Give a Hug to someone you know is RARE!
smile emoticon
By Dr. Bissell MD & Dr Bruce Wang & Jennifer Lai MD Overview of HCP & VP Facts
Friday - February 26, 2016 @ 10:30:21
Hereditary Coproporphyria
D Montgomery Bissell, MD, Bruce Wang, MD, and Jennifer Lai, MD.
Hereditary coproporphyria (HCP) is an acute (hepatic) porphyria in which the acute symptoms are neurovisceral and occur in discrete episodes. Attacks typically start in the abdomen with low-grade pain that slowly increases over a period of days (not hours) with nausea progressing to vomiting. In some individuals, the pain is predominantly in the back or extremities. When an acute attack is untreated, a motor neuropathy may develop over a period of days or a few weeks. The neuropathy first appears as weakness proximally in the arms and legs, then progresses distally to involve the hands and feet. Some individuals experience respiratory insufficiency due to loss of innervation of the diaphragm and muscles of respiration. Acute attacks are associated commonly with use of certain medications, caloric deprivation, and changes in female reproductive hormones. About 20% of those with an acute attack also experience photosensitivity associated with bullae and skin fragility.
Diagnosis/testing.
The most sensitive and specific biochemical screening test for any one of the acute porphyrias (including HCP) during an acute attack is a striking increase in urinary porphobilinogen (PBG). Quantitative analysis of porphyrins in both urine and feces is essential to distinguish between the different acute porphyrias and establish the diagnosis of HCP. Identification of a heterozygous pathogenic variant in CPOX (encoding the enzyme coproporphyrinogen-III oxidase) confirms the diagnosis and enables family studies.
Management.
Treatment of manifestations: Acute attacks are treated by discontinuation of any medications thought to induce attacks, management of dehydration and/or hyponatremia, administration of carbohydrate, and infusion of hematin. Treatment of symptoms and complications should be with medications known to be safe in acute porphyria (see www.drugs-porphyria.org).
A minority of affected individuals experience repeat acute attacks, in which case management strategies include suppression of ovulation in females, prophylactic use of hematin, and liver transplantation when attacks and neurologic complications persist despite multiple courses of hematin.
Prevention of primary manifestations: Agents or circumstances that may trigger an acute attack (including use of oral contraception in women) are avoided. Suppression of menses using a GnRH agonist (leuprolide, nafarelin, and others) may help CPOX heterozygotes who experience monthly exacerbations.
Prevention of secondary complications: In CPOX heterozygotes undergoing surgery, intravenous glucose is provided in the perioperative period and non-barbiturate agents are used for induction of anesthesia.
Agents/circumstances to avoid: Fasting, use of female reproductive hormones, and certain drugs including barbiturates and phenytoin.
Evaluation of relatives at risk: If the family-specific CPOX pathogenic variant is known, clarification of the genetic status of relatives at risk allows early diagnosis of heterozygotes and education regarding how to avoid risk factors known to be associated with acute attacks.
Genetic counseling.
HCP is inherited in an autosomal dominant manner with reduced penetrance. Most individuals with HCP have an affected parent; the proportion with a de novo pathogenic variant is unknown. Each child of an individual with HCP has a 50% chance of inheriting the CPOX pathogenic variant. Because of reduced penetrance, many individuals with a CPOX pathogenic variant have no signs or symptoms of HCP. Prenatal diagnosis for pregnancies at increased risk is possible if the pathogenic variant in an affected family member is known.
Hereditary coproporphyria (HCP) is classified as both an acute (hepatic) porphyria (with neurologic manifestations that occur as discrete, severe episodes) and a chronic (cutaneous) porphyria with long-standing photosensitivity.
Suggestive Findings
Acute hepatic porphyriais suspected in individuals with the following symptoms or findings:
Nausea for at least 48 hours
Abdominal, back, or extremity pain for at least 48 hours
New-onset seizures
Hyponatremia Note: Although CPOX pathogenic variants occur equally in males and females, acute attacks are much more frequent in women, mainly between ages 16 and 45 years (the years of active ovulation).
Chronic cutaneous porphyria is suspected in individuals with bullae and fragility of light-exposed skin which result in depigmented scars; however, the cutaneous signs occur in only a minority of heterozygotes, even during an acute attack.
Establishing the Diagnosis
The diagnosis of HCP is established by either biochemical testing (Table 1) or identification of a heterozygous pathogenic variant in CPOX on molecular genetic testing (Table 2).
Biochemical Testing
For an individual with pain and neurologic signs, the initial goal is to determine if the symptoms can be attributed to an attack related to any one of the acute porphyrias (i.e., ALA dehydratase deficiency porphyria, acute intermittent porphyria, hereditary coproporphyria, or variegate porphyria) (see Differential Diagnosis). Note: Since initial management is the same for all four types of acute porphyria, it is not necessary to determine at the outset of treatment which one of the four types of acute porphyria is present.
The most sensitive and specific biochemical diagnostic tests for HCP are detailed in Table 1. Once the diagnosis of an acute porphyria is established by identification of a striking increase in urinary porphobilinogen (PBG), quantitative analysis of porphyrins in both urine and feces may help define the specific type (Figure 1).
Profile of heme precursor excretion for the types of hepatic porphyria. The pathway of heme synthesis (arrows) is served by a series of enzymes (boxes). Mutations that decrease the function (more)
Active HCP is suggested by a quantitative urinary porphobilinogen (PBG) that is at least threefold the upper limit of normal.
The characteristic finding in stool is COPRO >> PROTO, quantified as units/g dry weight of feces. Note: Some laboratories report units/24 hours, which is inherently inaccurate. US laboratories that do the more precise analysis include ARUP (Salt Lake City, UT) and the Porphyria Laboratory, University of Texas Medical Branch, K Anderson, MD, Director (Galveston, TX).
The diagnosis is further substantiated by analysis of the COPRO-III/COPRO-I fecal porphyrin ratio, showing that 60%-95% of the total COPRO is isomer-III. In a normal (or negative) test, the predominant fecal porphyrin is PROTO, and the COPRO isomer III/I ratio in many cases is <0.5 [Kühnel et al 2000].
Table 1.
Biochemical Characteristics of Hereditary Coproporphyria (HCP)
Deficient Enzyme
Urine
Stool
Active
Asx
Active
Asx
Coproporphyringen-III oxidase 1, 2
PBG 3, 4 COPRO 5
Normal PBG COPRO 6
COPRO >> PROTO 7
See footnote 8
Active = symptomatic CPOX heterozygotes
Asx = asymptomatic CPOX heterozygotes
PBG = porphobilinogen
NormaI PBG = <2 mg (0.85 μmol) per g urine creatinine
COPRO = coproporphyrin
PROTO = protoporphyrin
1.
Also known as coproporphyrinogen oxidase and coproporphyrinogen decarboxylase
2.
The enzyme assay is not widely available and is not used for diagnostic purposes
3.
Active HCP is suggested by a quantitative urine PBG that is at least 3-fold the upper limit of normal
4.
Commercial laboratories offer quantitative delta aminolevulinic acid (ALA), PBG, and fractionated urine porphyrins. Values normalized to urine creatinine are satisfactory for clinical use, making a 24-hour collection unnecessary.
5.
See Differential Diagnosis for discussion of nonspecific elevation of COPRO in the urine.
6.
Fractionated urine porphyrins may reveal a minor rise in COPRO (<3-fold the upper limit of normal); however, this is nonspecific and insufficient for diagnosis (see Differential Diagnosis).
7.
60%-95% of the total COPRO is isomer-III.
8.
Fecal porphyrin analysis is the best test for distinguishing HCP from nonspecific coproporphyrinuria: heterozygotes show a predominance of fecal COPRO and an elevated COPRO III/I ratio (see Biochemical Testing).
Molecular Genetic Testing
Molecular testing approaches start with single-gene testing targeting the type of acute porphyria suggested by biochemical testing.
Sequence analysis of CPOX is performed first, followed by gene-targeted deletion/duplication analysis if no pathogenic variant is found.
If detailed CPOX testing is negative, PPOX (the gene for variegate porphyria [VP]) is analyzed. The biochemical findings in HCP and VP can overlap, leading to misassignment of diagnosis in some instances.
Table 2.
Summary of Molecular Genetic Testing Used in Hereditary Coproporphyria
Gene 1
Test Method
Proportion of Probands with a Pathogenic Variant 2 Detectable by This Method
Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exonor whole-gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here.
4.
Sequence analysis identified a pathogenic variant in 29 of 31 (94%) individuals with the clinical and biochemical diagnosis of HCP [Whatley et al 2009].
5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Examples of methods that may be used include: quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and gene-targeted microarray designed to detect single-exon deletions or duplications.
Biallelic CPOX pathogenic variants. Certain CPOX pathogenic variants (notably D400-K404 in exon 6) in the homozygous state result in a disorder called harderoporphyria [Hasanoglu et al 2011]. Some pathogenic variants may affect the active site of the enzyme, possibly causing premature separation of CPOX from its substrate. The result is incomplete decarboxylation of COPROgen yielding the tricarboxylic intermediate harderoporphyrinogen instead of dicarboxylic PROTOgen (Figure 1).
Heterozygotes for a CPOX pathogenic variant that is associated with harderoporphyria have no symptoms; those with biallelic pathogenic variants have large amounts of harderoporphyrin in the stool and neonatal onset of hemolytic anemia and jaundice, which can resemble congenital erythropoietic porphyria, an autosomal recessive condition with hemolysis and severe photosensitivity in infancy. They can also have acute porphyric attacks with high levels of urinary ALA and PBG. One infant died at age five months, apparently from complications of an acute attack [Hasanoglu et al 2011]. Some develop normally while others exhibit short stature.
Hereditary coproporphyria (HCP) is classified as both an acute and chronic porphyria. Porphyrias with neurologic manifestations are considered acute, because the symptoms occur as discrete, severe episodes. Porphyrias with cutaneous manifestations are considered chronic, because photosensitivity is long standing (see Table 3).
In a German study of 46 individuals with acute HCP, 90% had abdominal pain; only 13% had cutaneous findings despite substantial overproduction of coproporphyrin [Kühnel et al 2000]. An earlier British study of 111 individuals with HCP reported similar findings [Brodie et al 1977].
Symptoms prior to puberty in individuals who are heterozygous for a CPOX pathogenic variant have never been observed.
Fertility and longevity do not appear to be reduced in CPOX heterozygotes.
Acute Attacks
The initial symptoms of an acute attack are nonspecific, consisting of low-grade abdominal pain that slowly increases over a period of days (not hours) with nausea progressing to vomiting of all oral intake.
Typically the pain is not well-localized but in some instances does mimic acute inflammation of the gallbladder, appendix, or other intra-abdominal organ. In most instances the abdominal examination is unremarkable except for diminished bowel sounds consistent with ileus, which is common and can be seen on abdominal radiography. Typically fever is absent. In a young woman of reproductive age, the symptoms may raise the question of early pregnancy.
Prior to the widespread use of abdominal imaging in the emergency room setting, some individuals with abdominal pain and undiagnosed acute porphyria underwent urgent exploratory surgery. Thus, a history of abdominal surgery with negative findings was considered characteristic of acute porphyria.
A minority of affected individuals has predominantly back or extremity pain, which is usually deep and aching, not localized to joints or muscle groups.
Neurologic manifestations. Seizures may occur early in an attack and be the problem that brings the patient to medical attention. In a young woman with abdominal pain and new-onset seizures, it is critical to consider acute porphyria because of the implications for seizure management (see Management).
When an attack is unrecognized as such or treated with inappropriate medications, it may progress to a motor neuropathy, which typically occurs many days to a few weeks after the onset of symptoms. The neuropathy first appears as weakness proximally in the arms and legs, then progresses distally to involve the hands and feet. Neurosensory function remains largely intact.
In some individuals the motor neuropathy eventually involves nerves serving the diaphragm and muscles of respiration. Ventilator support may be needed.
Tachycardia and bowel dysmotility (manifest as constipation) are common in acute attacks and believed to represent involvement of the autonomic nervous system.
Of note when the acute attack is recognized early and treated appropriately (see Management), the outlook for survival and eventual complete recovery is good.
Psychosis. The mental status of people presenting with an acute attack of porphyria varies widely and can include psychosis. Commonly, however, the predominant feature is distress (including pain) that may seem hysterical or feigned, given a negative examination, absence of fever, and abdominal imaging showing some ileus only. Incessant demands for relief may be interpreted as drug-seeking behavior.
Because of the altered affect in acute porphyria, it has been speculated that mental illness is a long-term consequence of an attack and that mental institutions may house a disproportionately large numbers of individuals with undiagnosed acute porphyria. Screening of residents in mental health facilities by urinary PBG and/or PBG deaminase activity in blood (which diagnoses acute intermittent porphyria) has been done, with mixed results [Jara-Prado et al 2000]. The experience of those who have monitored patients over many years suggests that heterozygotes who are at risk for one of the acute porphyrias are no more prone to chronic mental illness than the general population; however, a prospective study is needed.
Kidney and liver disease. In people with any type of acute porphyria, the kidneys and liver may develop chronic changes that often are subclinical. One manifestation of the liver problem is excess primary liver cancer (hepatocellular carcinoma), which arises mainly after age 60 years. This and the kidney disease may be restricted largely to heterozygotes with chronically elevated plasma or urine delta aminolevulinic acid (ALA).
Inasmuch as ALA and porphobilinogen (PBG) tend to be minimally elevated or normal in HCP heterozygotes, the risk of hepatic and renal complications may be less in HCP than in acute intermittent porphyria.
Circumstances commonly associated with acute attacks are caloric deprivation, changes in female reproductive hormones, and use of porphyria-inducing medications or drugs:
Caloric deprivation. Fasting appears to sensitize the heme-synthetic pathway to an inducer, which could be external (i.e., a medication) or internal (ovarian hormones). The sensitizing effect of caloric deprivation was demonstrated in the 1960s in experimental animals and has been confirmed by clinical observation. People who fail to eat because of intercurrent illness or who undertake drastic weight loss are predisposed to an acute attack. First attacks have been reported after reduction gastroplasty for obesity [Bonkovsky et al 2008]. CPOXheterozygotes undergoing surgery are at risk because of the routine preoperative fast. This and other anecdotal experience have led to consensus that the first line of treatment for an acute attack is intravenous glucose, which is occasionally helpful.
Changes in female reproductive hormones. A role for female reproductive hormones can be inferred from the fact that acute attacks are infrequent prior to menarche and after menopause. Some women have monthly attacks that appear a few days before the onset of menstruation (when progestins peak). Attacks have been linked to use of oral contraceptives; the risk may be associated more with the progesterone component than the estrogen component.
Chronic (cutaneous) manifestations. Photocutaneous damage is present in only a small minority of those with acute attacks. Bullae and fragility of light-exposed skin, in particular the backs of the hands, result in depigmented scars. Facial skin damage also occurs, with excess hair growth on the temples, ears, and cheeks; this is more noticeable in women than in men.
The cutaneous findings in HCP resemble those in porphyria cutanea tarda (PCT) and in variegate porphyria (VP).
Threshold for a pathogenic effect of porphyrins and their precursors. Clinically active acute porphyria is associated with substantial elevation of the precursors ALA and PBG in the blood and urine; the cutaneous porphyrias are associated with increased porphyrins in blood, urine, and feces. In the acute porphyrias and cutaneous porphyrias, a threshold for symptoms appears to exist.
Acute (hepatic) porphyrias. A threshold for acute attacks is suggested by the fact that in virtually all symptomatic individuals, urinary PBG excretion exceeds 25 mg/g creatinine, or more than tenfold the upper limit of normal. Urinary ALA excretion increases roughly in parallel.
In contrast, in asymptomatic individuals the baseline urinary PBG excretion varies widely, usually low or normal but occasionally exceeding 25 mg/g creatinine. For this reason, it is advisable to establish the baseline urinary PBG excretion for CPOX heterozygotes (see Management, Evaluations Following Initial Diagnosis).
Chronic (cutaneous) porphyrias. A threshold has been well defined for porphyria cutanea tarda (PCT), in which photosensitivity occurs at values of urine uroporphyrin (the predominant pathway intermediate) that are more than 20-fold the upper limit of normal. However, the same is not apparent with regard to urine coproporphyrin: only a minority of CPOX heterozygotes exhibit any photosensitivity.
Of note, in individuals with HCP and chronic liver disease the cutaneous component may be more prominent than expected for the observed urine or plasma PBG concentration. Coproporphyrin leaves the plasma largely via the liver going into bile. In chronic liver disease, bile transport processes or bile formation may be impaired, leading to accumulation of coproporphyrin in plasma, which then results in photosensitivity.
Pathophysiology
Although the bone marrow actively produces heme (for hemoglobin synthesis), the liver is the main source of precursors in the acute (hepatic) porphyrias: acute attacks are precipitated when environmental factors stimulate increased hepatic heme synthesis and the genetically altered step in heme production becomes rate-limiting (Figure 1). Heme synthesis in the liver largely serves production of the cytochrome P450 family of heme-proteins, which are present in high concentration in the liver and have a relatively high turnover rate.
It is estimated that 20%-25% of total heme production normally occurs in the liver [Billing 1978]; however, that proportion increases when the liver is exposed to xenobiotics that undergo oxidative metabolism and stimulate cytochrome production (especially CYP3A4).
Acute attacks. The precursors ALA and PBG, unlike porphyrins, are colorless and non-fluorescent and do not contribute to photosensitivity in porphyria. Rather, ALA and PBG are highly associated with the neurologic manifestations of acute porphyria and are probably causal, although the mechanism remains speculative. The currently favored hypothesis implicates ALA (more than PBG), in part because acute neurologic symptoms occur in two other inherited conditions involving overproduction of ALA but not PBG (delta aminolevulinic acid dehydratase deficiency porphyria (ADP) and tyrosinemia). In addition, lead poisoning causes a similar biochemical derangement by binding the sulfhydryls of ALA dehydratase and reducing enzymatic activity; the symptoms in lead poisoning closely mimic those of acute porphyria [Bissell et al 2015]. Experimental studies indicate that ALA is a pro-oxidant species that is capable of damaging the inner membrane of mitochondria [Vercesi et al 1994].
Organ transplantation has established that the liver is responsible for acute attacks. Liver transplantation has cured individuals with refractory acute symptoms [Soonawalla et al 2004]. Moreover, transplantation of a porphyric liver into a normal recipient resulted in high circulating levels of ALA and PBG and symptoms of porphyria [Dowman et al 2011].
Cutaneous manifestations. Porphyrins are energized by blue light (peak wave length 410 nm). In a test tube, as activated porphyrins relax back to the ground state the released energy is evident as red fluorescence (ca. 625 nm). In vivo, the cycle of light activation and relaxation back to the ground state causes tissue damage, the nature of which varies with the porphyrin. URO and COPRO give rise to bullae and fragility of light-exposed skin, in particular the backs of the hands.
Genotype-Phenotype Correlations
HCP.CPOX pathogenic variants are not clustered around the enzymatic site. Furthermore, no correlation exists between the clinical phenotype and the residual enzymatic activity measured in vitro for a given pathogenic variant [Lamoril et al 2001].
Double heterozygosity for pathogenic variants in genes causing two different types of acute (hepatic) porphyria.Double heterozygotes for a pathogenic variant in CPOX and either a pathogenic variant in PPOX (variegate porphyria[VP]) [van Tuyll van Serooskerken et al 2011] or ALAD (ALA dehydratase deficiency porphyria [ADP]) [Akagi et al 2006] have been described. The phenotypes of such double heterozygotes vary but are not necessarily more severe than those associated with heterozygosity for either pathogenic variant alone, suggesting that double heterozygotes for two different types of acute porphyria may not be as rare as has been assumed.
Penetrance
Because population studies to determine the prevalence of HCP heterozygosity have not been done, the penetrance ofCPOX pathogenic variants is unknown. Given the rarity of acute attacks of HCP relative to acute intermittent porphyria (AIP), it is suspected that only a small minority of CPOX heterozygotes express the clinical disease. In 32 members of an Australian family, 14 (including 10 adults) were determined to have HCP on the basis of a high fecal COPRO III/I ratio and/or low lymphocyte CPOX enzyme activity; however, only one had clinical symptoms of porphyria [Blake et al 1992].
HCP, along with AIP and VP, are genetic disorders with reduced penetrance. Heme production in most heterozygotes appears to be adequate for physiologic homeostasis. Thus, environmental or physiologic factors play a role in the pathogenesis of acute attacks (see Management, Agents/Circumstances to Avoid). Although genetic co-factors may also be involved, none has been identified to date.
Nomenclature
Coproporphyria describes urine with an elevated level of coproporphyrin of any cause.
Coproporphyria in individuals heterozygous for a CPOX pathogenic variant is referred to as hereditary coproporphyria.
Prevalence
Clinical experience suggests that HCP is the least prevalent of the three principal types of acute porphyria: AIP, VP, and HCP. However, symptoms in HCP may be less frequent than in AIP or VP. Population surveys for CPOX pathogenic variants have not been reported.
The genetic porphyrias comprise a group of distinct diseases, each resulting from alteration of a specific step in the heme synthesis pathway that results in accumulation of a specific substrate (Figure 1).
In Table 3 the porphyrias are grouped by their principal clinical manifestations (neurolovisceral or cutaneous) and the tissue origin of the excess production of pathway intermediates (liver [i.e., hepatic] or bone marrow [i.e., erythropoietic]).
Porphyrias with neurovisceral manifestations are considered acute because the symptoms occur as discrete, severe episodes, which may be spontaneous but frequently are induced by external factors. The four acute porphyrias are: ALA dehydratase-deficiency porphyria (ADP), acute intermittent porphyria (AIP), HCP, and variegate porphyria (VP). Only a few individuals with ADP have been reported in the world literature.
Porphyrias with cutaneous manifestations include either chronic blistering skin lesions (i.e., VP as well as PCT, HCP, CEP, and hepatoerythropoietic porphyria [HEP]) or acute non-blistering photosensitivity (i.e., EPP and XLP).
Porphyrias with neurovisceral manifestations have been considered acute in part because the most common of these disorders, named acute intermittent porphyria, is the prototype for the neurovisceral porphyrias in which symptoms can occur acutely as discrete, severe episodes; however, some affected individuals develop chronic manifestations, and a few remain susceptible to exacerbating factors throughout their lives.
2.
Photocutaneous manifestations of EPP are acute and non-blistering, in contrast to the chronic blistering in the other cutaneous porphyrias (including VP).
While these clinical distinctions are important for the differential diagnosis, biochemical analysis is always necessary; however, biochemical testing may fail to distinguish HCP from VP, in which case molecular genetic testing of CPOX(HCP) and PPOX (VP) may be the only definitive diagnostic test.
Lead intoxication. The predominant elevation of coproporphyrin that is characteristic of HCP can also be seen in lead intoxication, in which the symptoms resemble those of an acute porphyria. The additional diagnostic finding in heavy metal poisoning is elevation of ALA unaccompanied by any increase in PBG.
Rotor syndrome, inherited in an autosomal recessive manner and caused by simultaneous deficiencies of the organic anion transporting polypeptides OATP1B1 and OATP1B3, is also associated with coproporphyrinuria [van de Steeg et al 2012].
Nonspecific coproporphyrinuria. The most important differential diagnosis in an individual with elevated urine coproporphyrin is HCP vs nonspecific coproporphyrinuria. Of all the people referred to a porphyria center, the largest subgroup has nonspecific coproporphyrinuria. Elevation of urine coproporphyrin is associated with a wide range of clinical conditions. It is particularly frequent in acquired liver disease (e.g., chronic viral hepatitis), but can also be seen in neurologic or hematologic diseases. Rarely, it is caused by an inherited hepatic transporter defect.
Two tests helpful for the differential diagnosis of coproporphyrinuria are:
Urine PBG, which is more than tenfold elevated in the inherited acute porphyrias with active symptoms;
The ratio of copro-III to copro-I in feces as measured by high-performance liquid chromatography (used for fecal porphyrin fractionation in most commercial labs). In nonspecific coproporphyrinuria the ratio is usually similar to that in normal controls [Gibson et al 2000].
For a case example of misdiagnosis of nonspecific coproporphyrinuria, click here.
To establish the extent of disease and needs of an individual diagnosed with hereditary coproporphyria (HCP), the following evaluations are recommended in an individual with acute abdominal symptoms:
Detailed neurologic examination for signs of motor neuropathy (which indicates a more advanced attack and, therefore, the need for early treatment with hematin)
Inquiry into possibility of seizures
Measurement of serum sodium concentration. Hyponatremia is characteristic and may be profound (serum sodium concentration <110 mEq/L), requiring urgent correction with due regard for the risk of central pontine myelinolysis.
Quantitation of urinary excretion of porphobilinogen (PBG) on several occasions over a few months to establish a baseline for future use in determining if a new symptom or drug reaction is due to an acute attack (In an acute attack urinary excretion of PBG is substantially elevated over the baseline.)
Evaluate those with nausea and vomiting for dehydration and hyponatremia, which is characteristic and may be profound (serum sodium concentration <110 mEq/L), requiring urgent correction with due regard for the risk of central pontine myelinolysis.
Provide glucose-containing IV solution to reverse the fasting state. Note: Caution is indicated in patients with hyponatremia, as aggressive administration of dextrose in water may cause the serum sodium concentration to drop to a critically low level.
Treat seizures with a short-acting benzodiazepine (e.g., midazolam) or with magnesium, which has been used for eclamptic seizures [Sadeh et al 1991]. Note: A number of the commonly used anti-seizure medications, including phenytoin and sodium valproate, are contraindicated because of the risk of further exacerbating an attack (see Agents/Circumstances to Avoid).
Intravenous hematin is the treatment of choice in moderate to severe acute attacks. Order hematin at the time that an acute attack requires hospitalization. Although hematin is not stocked by most hospital pharmacies, it can be obtained by overnight express from the manufacturer (Panhematin, Recordati, 1-888-575-8344). An alternative in Europe and elsewhere is heme arginate (Normosang; not yet approved in the US).
Some individuals recover with a glucose infusion only; those who do not respond in 24 to 48 hours should receive intravenous hematin.
When signs of a motor neuropathy are present, hematin is given as soon as possible. Hematin given at the initial signs of motor neuropathy may halt its progression; however, it has no effect on established motor deficits, which are the result of axonal degeneration.
Hematin is reconstituted at the bedside as described in the package insert. Human albumin may be used in place of water (132 mL of a 25% albumin solution) to reduce the risk of a chemical phlebitis, which is the main side effect of hematin administration [Anderson et al 2006].
The infusion is started without delay, as hematin in solution decays rapidly [Goetsch & Bissell 1986].
The preparation is given into a large peripheral vein or via central line over 10-15 minutes so as to minimize the risk of phlebitis. The dose is weight based at 3-4 mg/kg; 200 mg once daily is appropriate for most individuals.
The following responses to hematin infusion can be expected:
Decrease in the urine concentration of PBG, the first sign, occurs after two doses.
Clinical improvement is seen after a total of three or four doses, and typically is dramatic, with no further need of narcotic analgesia [Bissell 1988].
The motor neuropathy of acute attacks, when it occurs, does not respond to hematin administration. Return of function requires axonal regeneration and takes many months. Although it can be complete, some individuals have residual wrist drop or foot drop.
Liver transplantation. The experience with liver transplantation in acute porphyria is growing, with accumulating evidence that transplantation is curative in selected severe cases [Singal et al 2014]. The status of the disease in candidates for liver transplantation must be well-documented biochemically: they must not have responded to multiple courses of hematin and must be demonstrating neurologic complications.
Chronic (Cutaneous) Manifestations
For low-grade chronic or seasonal cutaneous symptoms, the only effective current treatment is avoidance of sun/light, whether direct or through window glass. Damage is caused by blue light and long-wave ultraviolet light (UVA), both of which pass through window glass:
Sun protection using protective clothing such as long sleeves, gloves, and wide brimmed hats
Protective tinted glass for cars and windows to prevent exposure to blue light. Grey or smoke colored filters provide only partial protection.
Note: Topical sunscreens are not helpful because they block UVB light, not the blue light that causes porphyrin-related skin injury.
The association of cutaneous manifestations with severe attacks (in which porphyrins as well as ALA and PBG are markedly increased) suggests that the cutaneous, as well as the neurovisceral, symptoms could respond to hematin administration. Indeed, this is the finding of a recent case report of an individual with severe HCP who was given maintenance hematin [Ma et al 2011].
Other. For more prolonged control of seizures, the combination of gabapentin and propofol is effective and safe.
Prevention of Primary Manifestations
Prevention of acute attacksinvolves the following:
Molecular genetic testing of at-risk relatives to identify those heterozygous for the CPOX pathogenic variant identified in the proband
Education of CPOX heterozygotes regarding circumstances that may trigger an acute attack (See Clinical Description.)
Selection of appropriate contraception for females. Oral contraceptives (birth control pills) are risky and not recommended. The recommended method of birth control for HCP heterozygotes is an IUD plus a barrier (diaphragm and/or condom).
A copper-eluting IUD is theoretically the safest in porphyria.
The hormone-eluting variety may also be safe because the systemic increase in hormone is quite small; however, little information exists.
Suppression of menses using a GnRH agonist. Leuprolide, nafarelin, and other GnRH agonists may help CPOXheterozygotes who experience monthly exacerbations.
CPOX heterozygotes undergoing surgery are at increased risk for an acute attack because of the routine preoperative fast and the (former) use of barbiturate (thiopental) induction of anesthesia. Adherence to the following recommendations greatly reduces the risk of an acute attack:
Minimizing the preoperative fast as much as possible and providing intravenous glucose (10% dextrose in half-normal saline) in the perioperative period
Anesthesia induction using non-barbiturate agents that have little or no P450-inducing activity (e.g., propofol, ketamine, short-acting benzodiazepines). Inhalation agents (isoflurane) and muscle relaxants also appear to be low-risk for triggering an attack.
Prevention of acute attacksdoes notinvolve the following:
Use of glucose. Because glucose is used to treat acute attacks, its use in preventing attacks has been suggested, and is in fact touted in lay discussions of porphyria; however, there is no evidence that heterozygotes can protect themselves by overeating or adopting a high-carbohydrate diet, and they risk becoming obese. Heterozygotes should adhere to a healthful diet with the usual balance of protein, fat, and carbohydrate. Weight loss is possible but only by incremental restriction of calories combined with exercise. Extreme diets (e.g., all bacon, all brown rice, starvation) are risky and should be avoided.
Liver transplantation. Because the vast majority of attacks respond to hematin and other supportive measures, liver transplantation has no role in prevention of acute attacks in a CPOXheterozygote.
Surveillance
For those who have chronically elevated ALA (which is infrequent in those who are asymptomatic) and/or are older than age 60 years, an annual check of liver and kidney function is recommended.
Current noninvasive techniques for assessment of fibrosis in the liver include transient elastography (FibroScan) and a blood-based test (FibroTest or FibroSure). Note: They have been vetted mainly for people with chronic viral hepatitis or steatohepatitis but may also be useful in porphyria.
For anyone with evidence of chronic liver injury, annual screening for hepatocellular carcinoma with abdominal imaging (such as ultrasound) and serum alpha-fetoprotein is indicated.
Agents/Circumstances to Avoid
Avoid the following:
Extreme caloric deprivation (i.e., total fasting, gastric bypass surgery)
Female reproductive hormones. Birth-control pills are risky and not recommended. For recommendations regarding contraception, see Prevention of Primary Manifestations, Selection of appropriate contraception for females.
Medications. Some drugs are clearly unsafe for CPOX heterozygotes. It is important to note, however, that many drugs are safe, lest providers regard individuals with acute porphyria as untreatable. Compilations of safe and unsafe drugs are available online and are updated as new information becomes available. Seewww.porphyriafoundation.com and www.porphyria-europe.com.
In theory, the most dangerous medications are inducers of CYPs, such as barbiturates and the related compound, phenytoin.
Evaluation of Relatives at Risk
It is appropriate to evaluate relatives at risk for HCP in order to identify as early as possible those who would benefit from education regarding the risk factors associated with acute attacks.
If the CPOX pathogenic variant in the family is known, molecular genetic testing can be used to clarify the genetic status of at-risk relatives.
If the CPOX pathogenic variant in the family is not known, a first-degree relative with symptoms can be evaluated with biochemical tests (see Diagnosis). Note: Although some CPOX heterozygotes have a diagnostic biochemical profile of heme precursors in urine and feces (see Table 1, Active columns), many have normal findings (see Table 1, Asymptomatic columns) and can only be diagnosed by molecular genetic testing.
The effect of pregnancy on inducing acute attacks is unpredictable. In general, serious problems during pregnancy are unusual. In fact, some women with recurrent symptoms associated with the menstrual cycle report improvement during pregnancy. Attacks, if they occur, are usually in the first trimester. The women most at risk are those with hyperemesis gravidarum and inadequate caloric intake [Aggarwal et al 2002]. Among antiemetics, ondansetron is not expected to precipitate or exacerbate acute attacks or to increase the risk for congenital anomalies in the fetus; however, metoclopramide should be avoided, as it may precipitate acute attacks [Shenhav et al 1997].
The experience with administration of hematin (or heme arginate, which is not available in the US) during pregnancy is limited. Badminton & Deybach [2006] published an anecdotal report of successful heme arginate treatment (without adverse fetal effect) in several women experiencing attacks of variegate porphyria or other acute porphyrias during pregnancy. Based on the absence of reported adverse effects, use of hematin to control exacerbations of acute intermittent porphyria during pregnancy has been recommended [Isenschmid et al 1992, Farfaras et al 2010].
Therapies Under Investigation
Due to the rarity of symptomatic acute porphyria, the efficacy of intravenous hematin has never been documented in a controlled trial. Efforts to accomplish this are under way.
Planned studies focus on individuals who have spontaneous acute attacks in the absence of any known external trigger in order to identify genetic cofactors that may be involved in such attacks, and are thus potential targets for new therapies.
Search ClinicalTrials.gov for access to information on clinical studies for a wide range of diseases and conditions.
Genetic counseling is the process of providing individuals and families with information on the nature, inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members. This section is not meant to address all personal, cultural, or ethical issues that individuals may face or to substitute for consultation with a genetics professional. ED.
Mode of Inheritance
Hereditary coproporphyria (HCP) is inherited in an autosomal dominant manner.
Risk to Family Members
Most individuals with HCP have an affected parent.
A proband with HCP may harbor a de novoCPOX pathogenic variant. The proportion of HCP caused by a de novo pathogenic variant is unknown.
Recommendations for the evaluation of parents of a proband with an apparent de novo pathogenic variant includemolecular genetic testing for the CPOX pathogenic variant identified in the proband.
Evaluation of parents may determine that one has a CPOX pathogenic variant but has not been previously diagnosed because of reduced penetrance. Therefore, an apparently negative family history cannot be confirmed until molecular genetic testing has been performed.
The risk to the sibs of the proband depends on the genetic status of the probands parents.
If a parent of the proband is heterozygous for the CPOX pathogenic variant identified in the proband, the risk to the sibs of inheriting the CPOX pathogenic variant is 50%. Because of reduced penetrance, many individuals heterozygous for a CPOX pathogenic variant do not manifest signs and symptoms of HCP.
If the CPOX pathogenic variant found in the proband cannot be detected in either parent, the risk to sibs is low but greater than that of the general population because of the possibility of germline mosaicism.
Offspring of a proband. Each child of an individual with HCP has a 50% chance of inheriting the CPOX pathogenic variant. Because of reduced penetrance, many individuals with a CPOX pathogenic variant do not manifest signs and symptoms of HCP.
Other family members
The risk to other family members depends on the status of the proband's parents.
If a parent is heterozygous for a CPOX pathogenic variant, his or her family members may be at risk.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Considerations in families with an apparent de novo pathogenic variant. When neither parent of a proband with HCP has the CPOX pathogenic variant or clinical evidence of the disorder, the CPOX pathogenic variant is likely de novo. However, non-medical explanations including alternate paternity or maternity (e.g., with assisted reproduction) or undisclosed adoption could also be considered.
Testing of at-risk asymptomatic relatives of individuals with HCP is possible after molecular genetic testing has identified the specific CPOX pathogenic variant in the family. Such testing should be performed in the context of formalgenetic counseling. The results of molecular genetic testing are not useful in predicting age of onset, severity, or specific symptoms. Note: Although some CPOX heterozygotes have a diagnostic biochemical profile of heme precursors in urine and feces (see Table 1, Active columns), many have normal biochemical test results (see Table 1, Asymptomatic columns) and can be diagnosed only by molecular genetic testing.
Family planning
The optimal time for determination of genetic risk and discussion of the availability of prenatal testing is before pregnancy.
It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
DNA banking is the storage of DNA (typically extracted from white blood cells) for possible future use. Because it is likely that testing methodology and our understanding of genes, allelic variants, and diseases will improve in the future, consideration should be given to banking DNA of affected individuals.
Prenatal Testing
If the CPOX pathogenic variant has been identified in an affected family member, prenatal testing for pregnancies at increased risk may be available from a clinical laboratory that offers either testing of this gene or custom prenatal testing.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the purpose is pregnancy termination rather than early diagnosis. Parents are encouraged to seekgenetic counseling before reaching a decision on the use of prenatal testing.
Note: The presence of a CPOX pathogenic variant detected by prenatal testing does not predict whether individuals will be symptomatic, or if they are, what the severity of the clinical manifestations will be. It is common for the offspring who inherit the CPOX pathogenic variant from a severely affected individual to be completely asymptomatic.
Preimplantation genetic diagnosis (PGD) may be an option for some families with a genetic disorder for which the pathogenic variant is known.
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. ED.
Data are compiled from the following standard references: gene from HGNC; chromosome locus, locus name, critical region, complementation group from OMIM; protein from UniProt. For a description of databases (Locus Specific, HGMD) to which links are provided, click here.
Gene structure.CPOX comprises seven exons; the reference sequence of the transcript is NM_000097.5. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic allelic variants. Pathogenic variants in all seven exons have been identified in persons with HCP (Human Gene Mutation Database, updated from Rosipal et al [1999]). They include missense and nonsense variants, small deletions, insertions, indels, splice variants, and large deletions.
Normal gene product. Coproporphyringen-III oxidase (synonyms: coproporphyrinogen oxidase and coproporphyrinogen decarboxylase), the product of CPOX, performs an oxidative decarboxylation without a metal, reducing agents, or obligatory cofactors. The details of this unusual reaction remain to be elucidated. The crystal structure of human coproporphyringen-III oxidase points to a dimer as the catalytically active unit.
Abnormal gene product. Some CPOX pathogenic variants likely alter enzyme activity by disrupting dimer formation [Lee et al 2005].
The 110-residue N-terminal segment is responsible for targeting the enzyme to mitochondria and is the site of its action on the substrate, coproporphyrinogen. Pathogenic variants in this region may affect translocation of the protein and, thus, reduce enzymatic function in tissues without changing activity in cell extracts.
Aggarwal N, Bagga R, Sawhney H, Suri V, Vasishta K. Pregnancy with acute intermittent porphyria: a case report and review of literature. J Obstet Gynaecol Res. 2002;28:1602. [PubMed]
Akagi R, Inoue R, Muranaka S, Tahara T, Taketani S, Anderson KE, Phillips JD, Sassa S. Dual gene defects involving delta-aminolaevulinate dehydratase and coproporphyrinogen oxidase in a porphyria patient. Br J Haematol. 2006;132:23743. [PubMed]
Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, Desnick RJ. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142:43950.[PubMed]
Anderson KE, Bonkovsky HL, Bloomer JR, Shedlofsky SI. Reconstitution of hematin for intravenous infusion.Ann Intern Med. 2006;144:5378. [PubMed]
Badminton MN, Deybach JC. Treatment of an acute attack of porphyria during pregnancy. Eur J Neurol.2006;13:6689. [PubMed]
Barbaro M, Kotajärvi M, Harper P, Floderus Y. Identification of an AluY-mediated deletion of exon 5 in the CPOX gene by MLPA analysis in patients with hereditary coproporphyria. Clin Genet. 2012;81:24956.[PubMed]
Billing BH. Twenty-five years of progress in bilirubin metabolism (1952-77). Gut. 1978;19:48191. [PMC free article] [PubMed]
Bissell DM. Treatment of acute hepatic porphyria with hematin. J Hepatol. 1988;6:17. [PubMed]
Bissell DM, Lai JC, Meister RK, Blanc PD. Role of delta-aminolevulinic acid in the symptoms of acute porphyria. Am J Med. 2015;128:3137. [PMC free article] [PubMed]
Blake D, McManus J, Cronin V, Ratnaike S. Fecal coproporphyrin isomers in hereditary coproporphyria. Clin Chem. 1992;38:96100. [PubMed]
Bonkovsky HL, Siao P, Roig Z, Hedley-Whyte ET, Flotte TJ. Case records of the Massachusetts General Hospital. Case 20-2008. A 57-year-old woman with abdominal pain and weakness after gastric bypass surgery. N Engl J Med. 2008;358:281325. [PubMed]
Brodie MJ, Thompson GG, Moore MR, Beattie AD, Goldberg A. Hereditary coproporphyria. Demonstration of the abnormalities in haem biosynthesis in peripheral blood. Q J Med. 1977;46:22941. [PubMed]
Dowman JK, Gunson BK, Bramhall S, Badminton MN, Newsome PN. Liver transplantation from donors with acute intermittent porphyria. Ann Intern Med. 2011;154:5712. [PubMed]
Farfaras A, Zagouri F, Zografos G, Kostopoulou A, Sergentanis TN, Antoniou S. Acute intermittent porphyria in pregnancy: a common misdiagnosis. Clin Exp Obstet Gynecol. 2010;37:25660. [PubMed]
Gibson PR, Grant J, Cronin V, Blake D, Ratnaike S. Effect of hepatobiliary disease, chronic hepatitis C and hepatitis B virus infections and interferon-alpha on porphyrin profiles in plasma, urine and faeces. J Gastroenterol Hepatol. 2000;15:192201. [PubMed]
Goetsch CA, Bissell DM. Instability of hematin used in the treatment of acute hepatic porphyria. N Engl J Med.1986;315:2358. [PubMed]
Hasanoglu A, Balwani M, Kasapkara CS, Ezgü FS, Okur I, Tümer L, Cakmak A, Nazarenko I, Yu C, Clavero S, Bishop DF, Desnick RJ. Harderoporphyria due to homozygosity for coproporphyrinogen oxidase missense mutation H327R. J Inherit Metab Dis. 2011;34:22531. [PMC free article] [PubMed]
Isenschmid M, König C, Fässli C, Haenel A, Hänggi W, Schneider H. Acute intermittent porphyria in pregnancy: glucose or hematin therapy? Schweiz Med Wochenschr. 1992;122:17415. [PubMed]
Jara-Prado A, Yescas P, Sánchez FJ, RÃos C, Garnica R, Alonso E. Prevalence of acute intermittent porphyria in a Mexican psychiatric population. Arch Med Res. 2000;31:4048. [PubMed]
Kühnel A, Gross U, Doss MO. Hereditary coproporphyria in Germany: clinical-biochemical studies in 53 patients. Clin Biochem. 2000;33:46573. [PubMed]
Lamoril J, Puy H, Whatley SD, Martin C, Woolf JR, Da Silva V, Deybach JC, Elder GH. Characterization of mutations in the CPO gene in British patients demonstrates absence of genotype-phenotype correlation and identifies relationship between hereditary coproporphyria and harderoporphyria. Am J Hum Genet.2001;68:11308. [PMC free article] [PubMed]
Lee DS, Flachsová E, Bodnárová M, Demeler B, Martásek P, Raman CS. Structural basis of hereditary coproporphyria. Proc Natl Acad Sci U S A. 2005;102:142327. [PMC free article] [PubMed]
Ma E, Mar V, Varigos G, Nicoll A, Ross G. Haem arginate as effective maintenance therapy for hereditary coproporphyria. Australas J Dermatol. 2011;52:1358. [PubMed]
Rosipal R, Lamoril J, Puy H, Da Silva V, Gouya L, De Rooij FW, Te Velde K, Nordmann Y, Martà sek P, Deybach JC. Systematic analysis of coproporphyrinogen oxidase gene defects in hereditary coproporphyria and mutation update. Hum Mutat. 1999;13:4453. [PubMed]
Sadeh M, Blatt I, Martonovits G, Karni A, Goldhammer Y. Treatment of porphyric convulsions with magnesium sulfate. Epilepsia. 1991;32:7125. [PubMed]
Schmitt C, Gouya L, Malonova E, Lamoril J, Camadro JM, Flamme M, Rose C, Lyoumi S, Da Silva V, Boileau C, Grandchamp B, Beaumont C, Deybach JC, Puy H. Mutations in human CPO gene predict clinical expression of either hepatic hereditary coproporphyria or erythropoietic harderoporphyria. Hum Mol Genet. 2005;14:308998. [PubMed]
Shenhav S, Gemer O, Sassoon E, Segal S. Acute intermittent porphyria precipitated by hyperemesis and metoclopramide treatment in pregnancy. Acta Obstet Gynecol Scand. 1997;76:4845. [PubMed]
Singal AK, Parker C, Bowden C, Thapar M, Liu L, McGuire BM. Liver transplantation in the management of porphyria. Hepatology. 2014;60:10829. [PMC free article] [PubMed]
Soonawalla ZF, Orug T, Badminton MN, Elder GH, Rhodes JM, Bramhall SR, Elias E. Liver transplantation as a cure for acute intermittent porphyria. Lancet. 2004;363:7056. [PubMed]
Stein P, Badminton M, Barth J, Rees D, Stewart MF., British and Irish Porphyria Network. Best practice guidelines on clinical management of acute attacks of porphyria and their complications. Ann Clin Biochem.2013;50:21723. [PubMed]
van de Steeg E, Stránecký V, Hartmannová H, Nosková L, HÅ?ebÃÄ?ek M, Wagenaar E, van Esch A, de Waart DR, Oude Elferink RP, Kenworthy KE, Sticová E, al-Edreesi M, Knisely AS, Kmoch S, Jirsa M, Schinkel AH. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J Clin Invest. 2012;122:51928. [PMC free article] [PubMed]
Vercesi AE, Castilho RF, Meinicke AR, Valle VG, Hermes-Lima M, Bechara EJ. Oxidative damage of mitochondria induced by 5-aminolevulinic acid: role of Ca2+ and membrane protein thiols. Biochim Biophys Acta. 1994;1188:8692. [PubMed]
Whatley SD, Mason NG, Woolf JR, Newcombe RG, Elder GH, Badminton MN. Diagnostic strategies for autosomal dominant acute porphyrias: retrospective analysis of 467 unrelated patients referred for mutational analysis of the HMBS, CPOX, or PPOX gene. Clin Chem. 2009;55:140614. [PubMed]
The work has been carried out under the auspices of the Porphyria Rare Disease Clinical Research Consortium (KE Anderson, Galveston, TX; DM Bissell, San Francisco, CA; JR Bloomer, Birmingham AL; HL Bonkowsky, Charlotte, NC; RJ Desnick, New York, NY; and JD Phillips, Salt Lake City, UT). The Consortium receives essential support from the NIH/NIDDK (1 U54 DK083909) and the American Porphyria Foundation.
Author History
D Montgomery Bissell, MD (2012-present) Theora Cimino, BS; University of California San Francisco (2012-2015) Jennifer Lai, MD (2012-present) Bruce Wang, MD (2012-present)
What is EPP & XLP Medical Facts
Wednesday - February 24, 2016 @ 10:30:12
Learn More - Overview of the Porphyrias
The Cutaneous Porphyrias
Cutaneous porphyrias primarily affect the skin. Areas of skin exposed to the sun become fragile and blistered, which can lead to infection, scarring, changes in skin coloring (pigmentation), and increased hair growth. Cutaneous porphyrias include congenital erythropoietic porphyria, erythropoietic protoporphyria and X-linked protoporphyria, porphyria cutanea tarda, and hepatoerythropoietic porphyria.
Erythropoietic Protoporphyria (EPP) and X-Linked Protoporphyria (XLP)
What are Erythropoietic Protoporphyria and X-Linked Protoporphyria?
EPP and XLP are both protoporphyrias with similar symptoms and similar biochemical findings, but each is caused by a mutation in a different gene. Some people refer to XLP as a variant form of EPP. Symptoms usually first occur in early childhood, and include sun sensitivity, marked by severe pain and swelling of sun-exposed areas, but typically with no blistering or scarring. EPP is caused by a deficiency of the enzyme, ferrochelatase (FECH), whereas XLP is due to an increased activity of another enzyme, aminolevulinic acid-synthase 2 (ALAS2). Both result in significant elevations of protoporphyrins in the liver, sometimes resulting in severe liver complications that are difficult to treat and sometimes require liver transplantation.
Who gets Erythropoietic Protoporphyria and X-Linked Protoporphyria?
EPP and XLP, combined, are the third most common porphyria, with an incidence of possibly 2 to 5 per 1,000,000, and the most common in children. EPP is an autosomal recessive disorder: an affected individual has either (1) a mutation on one of his / her FECH genes and a specific low-expression pre-disposing change (or alteration) on the other FECH gene or (2) a mutation on each of his / her FECH genes. XLP is an X-linked disorder, meaning that the disorder-causing gene, ALAS2, is on the X-chromosome (see Genetics 101). Because XLP is X-linked, males and females can both have symptoms, but males are usually more severely affected than females.
What causes Erythropoietic Protoporphyria and X-Linked Protoporphyria?
EPP is caused by a deficiency of the enzyme, ferrochelatase due to mutations in the FECH gene. XLP is caused by over-activity of amino-levulinic acid synthase due to a mutation in the ALAS2 gene. Both result in excess production of protoporphyrin in the bone marrow, transportation to the skin, and collection in the liver.
How is Erythropoietic Protoporphyria and X-Linked Protoporphyria diagnosed?
EPP and XLP are diagnosed by clinicalf symptoms and laboratory tests demonstrating increased protoporphyrin in red blood cells. EPP and XLP are differentiated by the levels of free- and zinc-complexed protoporphyrins: in EPP there is a predominance of free protoporphyrin over zinc protoporphyrin, whereas in XLP, the levels of free and zinc-complexed protoporphyrins are about equal.
What are treatments for Erythropoietic Protoporphyria and X-Linked Protoporphyria?
Treatment with pharmaceutical grade β-carotene (Lumitene, Tishcon) or cysteine may improve sunlight tolerance but does not lower porphyrin levels. Most patients must learn to avoid sunlight as much as possible. Deficiencies of iron and vitamin D should be prevented. To protect the liver from injury that might precipitate protoporphyric liver failure, Hepatitis A and B vaccinations are recommended, as is the avoidance of alcohol and other hepatotoxins.
Protoporphyric liver failure can appear suddenly and progress rapidly, and is generally treated with a combination of plasmapheresis, blood transfusion, hemin, cholestyramine, vitamin E, and ursodeoxycholic acid, which may reverse or delay liver damage. Levels of porphyrins in plasma and red blood cells should be followed closely during treatment. Liver transplantation is sometimes necessary. It is not yet possible to predict which patients will develop liver failure. Bone marrow transplantation is potentially curative in EPP and can prevent recurrent damage to the transplanted liver.
National Porphyria Awareness Week 2016
Monday - February 22, 2016 @ 10:30:11
National Porphyria Awareness Week 2016
This is the time of year that each of YOU has the opportunity to reach out to advance porphyria awarenss in your local and medical communities. YOU are key to spreading knowledge about porphyria. For example, the Cook family has enhanced awareness in Vernon, TX by hosting a Barrel Race and educating kids in school about EPP. Amanda Boston teaches her nurses and doctors about porphyria with her every hospitalization. Claire and Bob Sadowniczak manned the APF porphyria booth at the Hematology Convention. Louise Schlosser is hosting a patient support meeting. Amy Chapman writes a blog and Rob Saupe' assists with the APF website, Amy Rose Burke is a Facebook administrator. There are countless ways to enhance awareness. The APF will provide you with materials to help with your venue and will be glad to make suggestions to accomplish your goals if you need assistance. There are many ways to spread awareness in your own community, including the suggestions below:
Tell your story to the local television, radio or newspaper
Share your knowledge with doctors or at your hospital seminars
Set up exhibit booths at local health fairs or medical meetings
Assist the APF at national medical conventions
Volunteer your talents to help achieve the APF patient educational projects
Host a community race or car wash to benefit APF physician education
Volunteer your skills, like computer skills, or business acumen
Host a Fun Run or Walk-A-Thon or even a Night Run or Twilight Walk
Share your story in the Member Story Section of the APF website
Join Research Studies!
Friday - February 19, 2016 @ 10:30:01
Join Research Studies!
We are looking for patients volunteers to join The Longitudinal Study of the Porphyrias. Participation in this study is very easy. It does not require you to travel to the research site. The study involves a review of your medical records, completing comprehensive questionnaires about your symptoms and quality of life, and providing blood and urine samples. You will be working closely with porphyria experts and researchers.
The purpose of this long-term follow-up study is to provide a better understanding of the natural history of porphyrias, as affected by available therapies, and to aid in developing new forms of treatment.
We are also looking for volunteers to join A double-blind, randomized, placebo-controlled trial on the efficacy and safety of Panhematin. We are looking for patients with acute forms of porphyria: AIP, HCP or VP. This is a clinical trial, which means its purpose is to study an intervention or treatment. The study will occur in the hospital in Galveston, TX with planned follow up visits in the outpatient clinic or by telephone.
Patients are asked to come to the hospital as soon as possible after symptoms of an attack begin, because treatment is more effective if started early. Some patients, who have attacks that are regular and predictable, may come even before symptoms begin. The treatment the patient is given will be hidden (blinded) by using drapes and opaque containers so that participants and most of the staff do not know which treatment they are receiving. At least one pharmacist and one study nurse will know what treatment the participant is receiving, so we can be sure the treatment is properly prepared and safely administered.
Volunteering for research is the most important action you can take to change your future health because Research is the Key to Your Cure. You can be a Medical Hero and join many patients, who gave some of their time to help find important answers leading to new and improved treatments and a cure.
In order to participate in a study, please contact us at the APF 866-APF-3635. Or contact the research coordinator at the closest study center to you; their information can be found here:
Decreased nocturnal plasma melatonin levels in patients with recurrent acute intermittent porphyria attacks Medical Journal a must Read
Wednesday - February 17, 2016 @ 17:17:36
Abstract
Acute intermittent porphyria (AIP) is a hereditary disease characterized biochemically by a defect in the heme pathway enzyme porphobilinogen deaminase. There is wide variability in the neurologic clinical expression of AIP, and the disorder remains latent in most gene carriers. The natural history of the disease and results in a porphyric rat model sugget a significant relationship between tryptophan metabolites and clinical expression of the disease. In the present study, we examined urine and blood tryptophan metabolite levels in AIP women before, during and after acute attacks and treatment by heme arginate. Heme arginate treatment promptly decreased total tryptophan levels (from 69 ± 9, to 44 ± 5, mean ± SEM, μmole/l, p < 0.001), serotonin blood levels (from 629 ± 103, to 356 ± 80, nmole/l, p < 0.01) and the urinary excretion of 5-HIAA (from 3.9 ± 0.6, to 2.2 ± 0.4, μmole/mmole creatinine, p < 0.01). The plasma level of melatonin was found much lower than the normal control level at night (86.2 ± 70.3, vs the normal range, 409 ± 78.9, pmole/l ± SEM) and day time (38.8 ± 22.0, vs 75 ± 13.7). Heme arginate treatment did not influence melatonin levels. Our results support the involvement of abnormal tryptophan metabolism in the pathophysiology of AIP acute attacks. Low melatonin plasma levels in porphyric women suggest that the defect of the pineal hormone may be responsible for the recurrent aspect of porphyric attacks. A desynchronization of biological rhythms in AIP patients may increase the inducibility of hepatic ALA synthase to environmental risk factors and, specially, to sex steroid hormones.
open in overlay
Yves NORDMANN, Centre Français des Porphyries, Hôpital Louis Mourier, 92701 Colombes, France.
Clinuvel has been granted Orphan Drug Status to investigate Afamelanotide for VP, HCP and CEP. At present, we are awaiting FDA approval to treat EPP with Afamelanotide.
Clinuvel has been granted Orphan Drug Status to investigate Afamelanotide for VP, HCP and CEP. At present, we are awaiting FDA approval to treat EPP with Afamelanotide.
Keep your letters, emails, and photos coming.every day. We are making progress.
Read the full article below.
"Remember.Research is the key to your cure!"
Afamelanotide recognised as potential treatment for rare metabolic disorders
Clinuvel Pharmaceuticals Limited (ASX: CUV; XETRA-DAX: UR9; ADR: CLVLY) today announced that its lead drug SCENESSE (afamelanotide 16mg) has received an additional orphan drug designation (ODD) from the US Food and Drug Administration (FDA) for the treatment of cutaneous variants of porphyria. The ODD recognises the potential of afamelanotide to treat or prevent symptoms in rare forms of porphyria and offers incentives to Clinuvel to develop the drug for these patients.
SCENESSE (AFAMELANOTIDE 16MG) AND CUTANEOUS PORPHYRIAS
SCENESSE has been clinically evaluated as a photoprotective drug in one severe form of porphyria erythropoietic protoporphyria or EPP for which it has received marketing authorisation in Europe.1 It is proposed that SCENESSE may also have a photoprotective effect for patients with other rare forms of cutaneous porphyria: variegate porphyria (VP), hereditary coproporphyria (HCP) and congenital erythropoietic porphyria (CEP).
Porphyrias are a family of seven genetic metabolic disorders which cause malfunctions in the haem biosynthetic pathway. While classically grouped together, each of the five cutaneous porphyrias has clinically distinct symptoms, generally characterised as acute dermal reactions affecting the skin and anaphylactoid in nature which are caused by the accumulation and storage of phototoxic molecules (porphyrins) in the body.
When patients with cutaneous porphyria expose their skin to visible light both sunlight and certain artificial lights the porphyrins react and cause symptoms such as oedema and incapacitating deep burns leading to damage of soft tissue and eventually scarring. Patients are conditioned from childhood to recognise the source of their symptoms and withdraw from light and sun exposure to prevent phototoxicity, resulting in light starvation and social isolation.
SCENESSE has previously been granted orphan designation by the FDA for EPP and CEP. While no further trials are currently planned in cutaneous porphyrias, Clinuvel is working to make SCENESSE available to several patients with CEP on a name-patient basis due to the extreme severity and progressive character of this disorder. Only a few hundred cases of CEP have ever been recorded in the literature.
US ORPHAN DRUG DESIGNATION (ODD)
Orphan-drug designation is granted by the FDAs Office of Orphan Products Development to drugs which have the potential to diagnose or treat rare conditions, defined as those affecting less than 200,000 individuals in the United States. The additional orphan designation entitles Clinuvel to technical assistance throughout the development process for cutaneous porphyrias, potential fee reductions and tax credits, and seven years market exclusivity if approved for marketing by the FDA.
Beyond EPP we have always recognised that patients with other forms of cutaneous porphyria are deeply affected by their condition, Clinuvels Acting Chief Scientific Officer, Dr Dennis Wright said. The FDAs designation acknowledges the potential of afamelanotide to assist those patients.
- End -
1SCENESSE (afamelanotide 16mg) is approved as an orphan medicinal product in Europe for the prevention of phototoxicity in adult patients with EPP. Information on the product can be found on Clinuvels website at www.clinuvel.com.
About Clinuvel Pharmaceuticals Limited
Clinuvel Pharmaceuticals Ltd (ASX: CUV; XETRA-DAX: UR9; ADR: CLVLY) is a global biopharmaceutical company focused on developing drugs for the treatment of a range of severe disorders. With its unique expertise in understanding the interaction of light and human skin, the company has identified patient populations with a clinical need for photoprotection and for repigmentation. The worldwide prevalence of these patient groups range from 5,000 to 45 million. Clinuvels lead compound, SCENESSE (afamelanotide 16mg), was approved by the European Commission in 2014 for the prevention of phototoxicity in adult patients with erythropoietic protoporphyria (EPP).
Headquartered in Melbourne, Australia, Clinuvel has operations in Europe, Switzerland, the US and Singapore.
This release to the Australian Securities Exchange and to press may contain forward-looking statements, including statements regarding future results, performance or achievements. These statements involve known and unknown risks, uncertainties and other factors which may cause Clinuvels actual results, performance or achievements to be materially different from any future results, performances or achievements expressed or implied by the forward-looking statements. These statements reflect our current views with respect to future events and are based on assumptions and subject to risks and uncertainties. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Some of the factors that could affect the forward-looking statements contained herein include: that the FDA may require additional studies beyond the studies planned for product candidates or may not provide regulatory clearances, including for SCENESSE; that the FDA may not provide regulatory approval for any use of SCENESSE or that the approval may be limited; that Clinuvel may never file an NDA for SCENESSE regulatory approval in the US; that the Company may not be able to access adequate capital to advance its vitiligo programs; that the Company may not be able to retain its current pharmaceutical and biotechnology key personnel and knowhow for further development of its product candidates or may not reach favourable agreements with potential pricing and reimbursement agencies in Europe and the US.
Erythropoietic Protoporphyria (EPP) and X-Linked Protoporphyria (XLP)
Erythropoietic Protoporphyria (EPP) or Protoporphyria
Erythropoietic Protoporphyria is characterized by abnormally elevated levels of protoporphyrin IX in erythrocytes (red blood cells) and plasma (the fluid portion of circulating blood), and by sensitivity to visible light that is usually noticed in early childhood and occurs throughout life. EPP can result either from mutations of the ferrochelatase gene (FECH), or less commonly the delta-aminolevulinic acid synthase-2 gene (ALAS2). When EPP is due to an ALAS2 mutation it is termed X-linked protoporphyria (XLP), because that gene is found on the X chromosome.
Protoporphyrin accumulates first in the bone marrow in EPP, and then in red blood cells, plasma and sometimes the liver. Protoporphyrin is excreted by the liver into the bile, after which it enters the intestine and is excreted in the feces. It is not soluble in water so is not excreted in the urine.
EPP is the third most common type of porphyria, and the most common in childhood. It causes very painful photosensitivity and can greatly impair quality of life. Delay in diagnosis is greater than with any other type of porphyria.
Swelling, burning, itching, and redness of the skin may appear during or after exposure to sunlight, including sunlight that passes through window glass. This can cause mild to severe burning pain on sun-exposed areas of the skin. Usually, these symptoms subside in 12 to 24 hours and heal without significant scarring. Blistering and scarring are characteristic of other types of cutaneous porphyria but are unusual in EPP. Skin manifestations generally begin early childhood and are more severe in the summer.
There is an increased risk of gallstones, which contain protoporphyrin. Excess protoporphyrin can also cause liver damage. Less than 5% of EPP patients severe liver damage and a condition caused protoporphyric hepatopathy that sometimes requires liver transplantation.
Diagnosis and Genetic Counseling
EPP should be suspected in anyone with non-blistering photosensitivity especially when is it prolonged and beginning in childhood. It is easy to make a diagnosis, or rule it out, once it is suspected.
The diagnosis of EPP is established by finding an abnormally high level of total erythrocyte protoporphyrin, and showing that this increase is mostly free protoporphyrin rather than zinc protoporphyrin. There is considerable confusion about which test to order. Sometimes laboratories have measured only zinc protoporphyrin and reported results incorrectly as protoporphyrin or free erythrocyte protoporphyrin (FEP). Laboratories that measure total erythrocyte protoporphyrin, free protoporphyrin and zinc protoporphyrin and report results reliably are:
Porphyria Laboratory and Center, University of Texas Medical Branch at Galveston, 1-409-772-4661
Mayo Medical Laboratories, 1-800-533-1710
ARUP Laboratories
Porphyrins are almost always elevated in plasma in EPP, but may be normal in mild cases. Fecal porphyrins may be normal or increased.
An experienced biochemical laboratory can usually distinguish between patients with EPP and XLP, because the former have much less zinc protoporphyrin in their erythrocytes. This can be explained because in the marrow the enzyme ferrochelatase not only normally makes heme (iron protoporphyrin) from protoporphyrin and iron, but can also make zinc protoporphyrin, especially when excess protoporphyrin is present or iron is deficient. However, this does not replace DNA studies.
Rarely, EPP develops in adults in the presence of a bone marrow disorder such as polycythemia vera, and is due to expansion of a clone of red blood cell precursors in the marrow that is deficient in ferrochelase.
DNA studies are important for confirming the diagnosis of EPP and XLP and for genetic counseling. This should be completed first in a person known to have the disease, and the information about the mutations in that individual used to guide testing of family members.
When EPP is due to a FECH mutation the inheritance is described as autosomal recessive. It is most common to find that one severe mutation is inherited from one parent and another weak mutation inherited from the other parent. The weak mutation is quite common in normal Caucasians, rare in Blacks and even more common in Japanese and Chinese populations. This mutation is sometime referred to as hypomorphic because it results in formation of a less than normal amount of ferrochelatase. But is does not cause EPP unless it is paired with a severe mutation. The severe mutation is characteristic for an EPP family and is present in all affected individuals. Carriers of the severe mutation are not affected because they do not have the weak mutation. Affected individuals and unaffected carriers can transmit the severe mutation to the next generation. Some of their children will have EPP if the other parent has a copy of the weak mutation. Rarely, the weak mutation is absent in an EPP family and two severe mutations are found, with at least one producing some ferrochelatase.
In XLP, mutations of the ALAS2 gene, which is found on the X chromosome, causes an increase in the production of the enzyme ALAS2 in the bone marrow. Several of these gain of function mutations have been described in different XLP families. In XLP protoporphyrin production exceeds that needed for heme and hemoglobin formation. Like hemophilia and other X linked genetic diseases, XLP is more common in men. Women have two X chromosomes and are usually not affected because they have a normal as well as a mutated ALAS2 gene. Men have only one X chromosome and will be affected if they inherit an ALAS2 mutation. Women with an ALAS2mutation will, on average, pass that mutation to half of their daughters (who will usually be unaffected carriers) and to half of their sons (who will be affected).
Treatment and Management
1. Sunlight protection
Protection from sunlight is the mainstay of management of EPP, and this is necessary throughout life. Disease severity and porphyrin levels in erythrocytes and plasma probably remain high and relatively constant throughout life in EPP. However, this has been little studied and more longitudinal observations are needed. Life style, employment, travel and recreation require adjustment in order to avoid painful reactions to sunlight and even from exposure to fluorescent lighting. For these reasons EPP can substantially affect quality of life.
Protective clothing, including broad-brimmed hats, long sleeves, gloves and trousers (rather than shorts), is beneficial. Several manufacturers specialize on clothing made of closely woven fabrics for people with photosensitivity.
2. Beta-Carotene (Lumitene Tishcon)
Beta-carotene is an over the counter product that was originally developed in a purified form as a drug for the treatment of EPP, and was shown to be effective by Dr. Micheline Mathews-Roth at Harvard University and others. The pharmaceutical grade formulation is now distributed by Tishcon as Lumitene, and can be ordered by calling 1-800-866-0978 or via the website www.epic4health.com. Other products are less standardized and reliable and are not recommended.
Beta-carotene provides protection by quenching reactive oxygen products that form when protoporphyrin is activated in the skin by light. It is important to take an amount that is adequate to be protective. For more information about Lumitene, including a recommended dosing schedule, please see the Lumitene section of this website.
3. Other considerations
In an occasional patient, protoporphyrin causes liver problems, so monitoring liver function is important. EPP patients should also not use any drug or anesthetic which causes cholestasis (slowing down bile flow), and should also avoid alcohol. Women should avoid medications containing estrogen (birth-control pills, hormone replacement therapy), and men should avoid testosterone supplements, as these substances can also have deleterious effects on the liver of a person with EPP.
Consult a specialist. Because EPP is a rare condition, most physicians are not knowledgeable about it. Contact The American Porphyria Foundation, 713-266-9617 for contact with an expert and to provide further information. A Medic Alert bracelet with instructions to contact a specialist if needed is a worthwhile precaution.
Yearly monitoring. Testing to include erythrocyte total protoporphyrin, plasma porphyrin, complete blood counts, ferritin and liver function tests should be done yearly. Porphyry levels are expected to be stable and liver tests to remain normal. EPP patients may have evidence of iron deficiency, and an iron supplement may be advisable if the serum ferritin is below about 20 ng/mL.
Vitamin D. Because they avoid sunlight, EPP patients are likely to be deficient in vitamin D. A vitamin D supplement with calcium is recommended for bone health.
Liver protection. It is important to avoid other causes of liver disease that might promote the development of liver complications from EPP. Patients should avoid alcohol and other substances that might damage the liver, including many herbal preparations, and be vaccinated for hepatitis A and B.
Surgical lights. Strong operating room lights can cause photosensitivity of the skin and even surfaces of internal organs. Flexible membrane filters, such as CL5-200-X from Madico Co., are available to cover surgical lights and offer some protection. This is especially important in EPP patients with liver failure, which causes even greater increases in protoporphyrin levels and photosensitivity.
Drugs. Drugs that are harmful in other porphyrias are not known to make EPP worse, but are best avoided as a precaution. This may include estrogens and other drugs that might reduce bile formation. A short course of a non-steroidal anti-inflammatory drug can provide some pain relief after an episode of photosensitivity, but can cause ulcerations of the digestive track especially with prolonged use.
Laser treatment. According to Dr. Roth, laser treatments for hair removal or eye surgery have not been a problem in EPP people. But the doctor should be made aware of the diagnosis, and that laser output between 400 and 650 nanometers might be harmful. Before hair removal treatment, the doctor may irradiate a small area of the skin to be treated for the length of time it will take to do the hair removal to ascertain if the patient would react within the period of time that a reaction to sunlight would be expected in that patient.
Children with EPP. Avoiding sunlight can be difficult for children with EPP who have less sunlight tolerance than their friends. Camp Discovery is an option for such children. It provides a week-long summer camping experience of fishing, boating, swimming, water skiing, arts and crafts, and just plain fun for young people with skin disorders, and is sponsored by the American Academy of Dermatology. Full scholarships, including transportation, are provided by the American Academy of Dermatology through generous donations of their members and other organizations. Members of the Academy are asked to recommend candidates for Camp Discovery, so ask your child's doctor about sending your child to Camp Discovery.
Disneyland and Disney World are responsive to people with sun sensitivity. They will provide a pass to enable you to enter attractions without waiting in line in the sun.
Disneyland Go to "Town Hall" and explain your problem with photosensitivity. You should bring a physician's letter with you as well as an APF brochure explaining the type of Porphyria you have. The staff will ask you some questions about your limitations (e.g., whether or not you can climb stairs) and how many are in your group. Next time you return, be sure to bring the old card with you, as it will only take about half as long to go through the process on your next trip.
Disney World Proceed to the "Guest Relations" office at any park (Magic Kingdom, EPCOT, etc.) and request the Special Assistance Pass.
Remember to bring a doctor's note and explanation of your condition, because it is not necessarily visible. People on duty may not be familiar with light sensitivity and its consequences.
Research Opportunities
Patients with EPP and XLP can participate in the research through the Porphyrias Consortium. The American Porphyria Foundation has information on what research protocols are currently open.
·
The Porphyrias Consortium is conducting a Longitudinal Study to better define the natural history of the disease. This study is currently open for enrollment of new patients.
·
The Porphyrias Consortium will be starting a pilot study soon on a drug that may lower porphyrin levels in EPP.
·
Clinuvel Pharmaceuticals is developing afamelanotide (Scenesse) for the treatment of EPP. This drug is given by injection and increases skin pigmentation. Another study of this drug is expected to open within the next year.
All patients with porphyria are encouraged to enter the Porphyrias Registry at the Porphyrias Consortium website. A link to this website is found on the website of the American Porphyria Foundation. Registration demonstrates to NIH that patients and their families think that research on porphyrias is important. You can also ask that one of the 6 porphyria center in the Consortium contact you.
Write your Congressman EPP PATIENTS!!!
Friday - January 29, 2016 @ 10:00:17
Write your Congressman
Your 300 letters to the FDA regarding EPP were VERY effective. We are gaining ground! EPP is now on their radar. BUT we need your help. Now we need everyone to contact their congressmen and urge them to contact the FDA directly.
You may submit your letters via mail OR email. The website shown above provides you with all contact information. Below we have provided a template letter you may use, or, better yet, please write one of your own. The more letters, the better! Thank you all!
"Rememberresearch is the key to your cure!!"
[Insert Date]
The Honorable _________________
[Insert your Congressman's address here]
Dear Mr. ________,
As a person who has been diagnosed with Erythropoietic Protoporphyria (EPP), I am very concerned that the FDA has not approved Afamelanotide, a revolutionary drug to treat EPP. EPP is a rare, extremely photosensitive disease that causes severe burning of the skin, liver damage, Vitamin D deficiency and other major health problems. In other words, sunlight causes children and adults to suffer, and no one can avoid sunlight. When we discovered this drug would allow us to drive to work without pain, spend a day in the sun with our families and even do simple acts like walk to our cars without burning severely, we began to demand that the FDA approve Afamelanotide.
The EMA approved Afamelanotide in Europe in October 2014. In fact, almost half of worldwide patients can receive Afamelanotide for EPP, while US citizens have no FDA approval and continue to suffer. Instead, EPP patients in the US are forced to travel at great expense to purchase the drug in Switzerland. The demand has escalated to the point that people are asking for Congressional and media help. Recently, NBC, ABC, CNN, FOX, Telemundo and a host of other networks reported on EPP and this new treatment.
The American Porphyria Foundation, their Scientific Advisory Board and Porphyria Research Consortium, who performed the research, all recommend that the FDA approve Afamelanotide. As of yet, the FDA has not assessed the data from the clinical trials. Clinuvel, the drug manufacturer, cannot file a New Drug Application until the FDA assesses the data and allows their filing.
Afamelanotide has a long, safe and effective history. This timeline purports the readiness for this drug to receive accelerated approval.
â?¢ Trials for EPP started in 2006. Since then, there have been five clinical trials. In all other indications, including healthy volunteers, there have been more than 15 trials. More than 1,000 patients have received the drug. This is extremely important in that EPP is considered an ultra-rare disease, therefore, the numbers herein mean that a large part of the EPP population has been tested with the drug.
â?¢ Since 2006, a total of 146 patients continuously have been on Afamelanotide on a compassionate basis as part of the Special Access Schemes in Switzerland and Italy. The FDA has never allowed patients to have Afamelanotide on a compassionate basis.
â?¢ The European Medicines Agency (EMA) approved Afamelanotide for EPP on October 24, 2014. The FDA is keenly aware that Afamelanotide was approved in Europe as FDA representatives attended the plenary session in September 2014 before the EMA approval. The European Medicines Agency took record time to assess the novel and challenging nature of EPP and the unusual mode of action of the drug.
Most important to all of the EPP community is the FDA has not allowed trials for children. Every FDA delay adds five years to these children ever having access to the drug. While we await FDA approval of Afamelanotide, the FDA has approved treatments for wrinkle cream and double chin. Meanwhile, the EPP community suffers with severe pain and liver damage when a tiny implant of Afamelanotide would provide us with a normal life.
Please ask the FDA to grant accelerated approval for Afamelanotide.
Learn what could trigger symptoms. Talk to your doctor about the type of porphyria you have and become familiar with possible symptom triggers and ways to avoid them.
Inform your health care providers. Tell all your health care providers that you have porphyria. This is particularly important because sometimes treatments, medications or surgery can trigger porphyria symptoms.
Wear a medical alert bracelet or necklace. Have information about your condition inscribed on a medical alert bracelet or necklace, and always wear it.
Although there's no way to prevent porphyria, if you have the disease, these steps may help prevent symptoms:
Avoid medications known to trigger acute attacks. Ask your doctor for a list of safe and unsafe drugs.
Don't use alcohol or illegal drugs.
Avoid fasting and dieting that involves severe calorie restriction.
Don't smoke.
Minimize sun exposure. When you're outdoors, wear protective clothing and use a broad-spectrum sunscreen with a high sun protection factor (SPF).
Treat infections and other illnesses promptly.
Take steps to reduce emotional stress.
Because porphyria is an inherited disorder, your siblings and other family members may want to consider genetic testing to determine if they have the disease.
"Remember.Research is the key to your cure!"
Meeting with the FDA March 7!! IMPORTANT
Wednesday - January 27, 2016 @ 10:00:23
Meeting with the FDA March 7!!
The APF is meeting with the FDA on March 7, 2016 to discuss the ACUTE PORPHYRIAS (AIP, HCP, VP). We ask that you and your family members please join us at this meeting to share your story and your needs.
This is a very difficult meeting to gain. Please come help us explain the patient needs, problems and difficulties. Note this is not the EPP meeting we are trying to schedule but an acute porphyria meeting.
Please contact the APF if you need any further information or assistance.
Many signs and symptoms of porphyria are similar to those of other more common diseases. Also, because porphyria is rare, it can be more difficult to diagnosis. Lab tests are required to make a definitive diagnosis of porphyria and to determine which form of the disease you have.
If your doctor suspects porphyria, he or she may recommend these tests:
Urine test. If you have a form of acute porphyria, a urine test may reveal elevated levels of two substances: porphobilinogen (por-foe-bih-LIN-uh-jen) and delta-aminolevulinic (uh-me-no-lev-yoo-LIN-ik) acids, as well as other porphyrins.
Blood test. If you have a form of cutaneous porphyria, a blood test may show an elevation in the level of porphyrins in your blood plasma.
Stool sample test. Analysis of a stool sample may reveal elevated levels of some porphyrins that may not be detected in urine samples. This test may help your doctor determine your specific type of porphyria.
More tests may be needed to confirm the type of porphyria you have. Genetic testing may be suggested in the family of a person with porphyria.
Treatment depends on the type of porphyria you have and is directed at relieving symptoms.
Acute porphyrias
Treatment of acute porphyrias focuses on providing rapid treatment of symptoms and preventing complications. This may require hospitalization in severe cases. Treatment may include:
Stopping medications that may have triggered symptoms
Medication to control pain, nausea and vomiting
Prompt treatment of infections or other illness that may have caused symptoms
Intravenous sugar (glucose) or sugar taken by mouth, if able, to maintain an adequate intake of carbohydrates
Intravenous fluids to combat dehydration
Injections of hemin, a medication that is a form of heme, to limit the body's production of porphyrin
Cutaneous porphyrias
Treatment of cutaneous porphyrias focuses on reducing exposure to sunlight and the amount of porphyrins in your body to help eliminate your symptoms. This may include:
Drawing blood (phlebotomy). Drawing a certain amount of blood from one of your veins reduces the iron in your body, which decreases porphyrins. You may need to have a phlebotomy repeated at regular intervals before cutaneous porphyria goes into remission.
Medication. Drugs used to treat malaria hydroxychloroquine (Plaquenil) or, less often, chloroquine (Aralen) can absorb excess porphyrins and help your body get rid of them more quickly than usual. These medications are generally used only in people who can't tolerate a phlebotomy.
Beta carotene. Long-term treatment of cutaneous porphyrias may include daily doses of prescription beta carotene. Beta carotene may increase your skin's tolerance to sunlight. Your doctor can tell you what kind of beta carotene will work best for porphyria photosensitivity.
Reducing or eliminating triggers. Triggers, such as certain medications or too much sunlight, which activated the disease, should be reduced or removed if possible, with guidance from your doctor.
Vitamin D. Supplements may be recommended to replace vitamin D deficiency caused by avoidance of sunlight.
If you have signs and symptoms of porphyria, you're likely to start by seeing your primary care provider. However, because porphyria can be difficult to diagnose, you may be referred to a doctor who specializes in blood disorders (hematologist).
Here's some information to help you get ready, and what to expect from your doctor.
What you can do
Before your appointment, make a list of:
Any symptoms you're experiencing, including any that may seem unrelated to the reason for your appointment
Key personal information, including any major stresses or recent life changes
All medications, vitamins or other supplements that you're taking, including dosages
Preparing a list of questions before your appointment will help you make the most of your time. For porphyria, some basic questions to ask your doctor include:
What's the most likely cause of my symptoms?
What are other possible causes?
What kinds of tests do I need? Do I need genetic testing?
How severe is my condition?
What's the best course of action?
What are the alternatives to the primary approach that you're suggesting?
I have another health condition. How can I best manage these together?
Are there any dietary restrictions I need to follow?
What precautions do I need to take when spending time outdoors?
Do I need to be concerned about taking medications in the future?
Are there any brochures or other printed material that I can have? What websites do you recommend?
Should my family members be screened?
Will I need a medical alert bracelet?
Don't hesitate to ask any other questions during your appointment.
What to expect from your doctor
Your doctor is likely to ask you a number of questions, such as:
When did you first begin experiencing symptoms?
Have your symptoms been continuous or occasional?
How severe are your symptoms?
What, if anything, seems to improve your symptoms?
What, if anything, appears to worsen your symptoms?
Do any family members have similar symptoms?
"Remember.Research is the key to your cure!"
Risk Factors & Complications of All Porphyrias by Mayo Clinic
In addition to genetic risks, environmental factors may trigger the development of signs and symptoms in some types of porphyria. When exposed to the trigger, your body's demand for heme production increases. This overwhelms the deficient enzyme, setting in motion a process that causes signs and symptoms. Examples of triggers include:
Certain drugs (barbiturates or sulfonamide antibiotics or, less often, birth control pills, or some drugs that affect the mind or behavior, known as psychoactive drugs)
Chemicals
Dieting or fasting
Smoking
Physical stress, such as infections or other illnesses
Dehydration. Vomiting due to an attack of acute porphyria can lead to dehydration, which may require that you receive fluids through a vein (intravenously).
Breathing difficulties. Acute porphyrias can cause muscle weakness and paralysis, which can cause breathing problems. If left untreated, they can also lead to respiratory failure.
Low sodium in your blood. Called hyponatremia, this is usually linked to problems with sodium and water handling in your body.
High blood pressure. Porphyrin buildup can damage your kidneys and may result in high blood pressure (hypertension).
Chronic kidney failure. Porphyrin buildup may cause your kidneys to gradually lose their ability to function.
Liver damage. Some forms of porphyria cause excessive porphyrins in your liver, which may lead to severe liver damage that can eventually require a liver transplant.
Permanent skin damage. When your skin heals after cutaneous porphyria, it may have an abnormal appearance and coloring. Scars may remain on your skin as well, and lasting skin problems may cause your hair to fall out.
Porphyria is most often an inherited mutation in one of the genes involved in heme production, although environmental factors can trigger symptoms in some cases.
Heme is a major component of hemoglobin, the protein in red blood cells that carries oxygen from your lungs to all parts of your body. Heme also plays a role in breaking down chemicals so they can be removed from your body. Heme is made mainly in the bone marrow and liver through the production of porphyrin and linkage with iron.
Eight different enzymes add and convert natural, smaller building blocks into porphyrin, which becomes heme with the addition of iron. Deficiency of a specific enzyme that's involved in the body's process for making heme can result in the buildup of porphyrins, causing symptoms. Each type of porphyria is due to the deficiency of a different enzyme.
Genetics
Most forms of porphyria are inherited. Porphyria can occur if you inherit:
A defective gene from one of your parents (autosomal dominant pattern)
Defective genes from both parents (autosomal recessive pattern)
Just because you have inherited a gene or genes that can cause porphyria doesn't mean that you'll have signs and symptoms. You might have what's called latent porphyria, and never have symptoms. This is the case for most carriers of the abnormal genes.
"Remember.Research is the key to your cure!"
Symptoms of each type of Porphyria from Mayo Clinic
There are two general categories of porphyria acute, which mainly affects the nervous system, and cutaneous, which mainly affects the skin. Some types of porphyria have both nervous system symptoms and skin symptoms, and others have mainly one or the other.
Acute porphyrias
Acute porphyrias include forms of the disease that typically cause nervous system symptoms, which appear quickly and can be life-threatening. Acute porphyria attacks are rare before puberty and after menopause in women. Symptoms may last one to two weeks and usually improve slowly after the attack.
Possible signs and symptoms of acute porphyria include:
Severe abdominal pain
Swelling of the abdomen (abdominal distention)
Pain in your chest, legs or back
Constipation or diarrhea
Vomiting
Insomnia
Heartbeat you can feel (palpitations)
High blood pressure
Anxiety or restlessness
Seizures
Mental changes, such as confusion, hallucinations, disorientation or paranoia
Breathing problems
Muscle pain, tingling, numbness, weakness or paralysis
Red or brown urine
Cutaneous porphyrias
Cutaneous porphyrias include forms of the disease that cause skin symptoms as a result of oversensitivity to sunlight, but these forms don't usually affect your nervous system. Attacks may last for several days. With some forms, signs and symptoms may start during infancy or childhood.
As a result of sun exposure, you may experience:
Sensitivity to the sun and sometimes artificial light, causing burning pain
Sudden painful skin redness (erythema) and swelling (edema)
Blisters that take weeks to heal
Itching
Fragile skin
Scars or skin color changes from healing blisters
Increased hair growth
Red or brown urine
When to see a doctor
Many signs and symptoms of porphyria are similar to those of other, more common conditions. This can make it difficult to know if you're having an attack of porphyria. Any of the following symptoms should prompt you to seek immediate medical attention:
Severe abdominal pain, but sometimes pain in your chest, legs or back, accompanied by constipation, vomiting and sometimes diarrhea
Sensitivity to the sun and sometimes artificial light, causing burning pain and sudden painful skin blistering, redness (erythema) and swelling (edema)
Red or brown urine
"Remember.Research is the key to your cure!"
Save the Date for the Patient Educational Meeting
Wednesday - January 6, 2016 @ 10:30:02
Save the Date for the Patient Educational Meeting
You are invited to a patient educational meeting in Houston, TX.
* Presentations by World Renowned Porphyria Experts.
* Meeting with the APF's Executive Director Desiree Lyon
* Opportunity to Participate in a Q & A Session.
* Meet Friends who Share Your Experiences with Porphyria.
* View the Latest Educational Material from the American Porphyria Foundation.
Date and Time: Monday, March 14, 2016, 6:30 PM CT, Houston, TX
Its bound to happen with the holidays, travel, family food and friends. We get sick bad and cold weather out, gotta go to work, take care of the family, but what about me? How do I take care of myself? Here are some great tips.
Hollys great post on Feel Better Day reminded me how its so easy when were illeither from a passing cold or a long-term diseaseto slip into the emotional dumps. But its not necessary to feel blue while your body is healing. In fact, many studies show that the brighter your mood the faster you mend. As someone who has had four colds this season, gets migraines sometimes, and has survived a bout of cancer and chemo, Ive come up with ways to stay up when my bodys down. Hopefully youll find them helpful too.
1) Remember: Your Body Is Sick, Not Your Brain. Im all for deepening the mind-body connection, but I find its crucial to let the stronger one lead the dance. I remind myself that Im not required to feel sad just because Im sick and delineate a happy divide between physical aches and pains and emotional ones. You can be in pain and not sufferor at least not add to the external suffering with a poor me monologue in your head. My fourth grade teacher Betsy blew my mind when I hurt my finger one day and she told me to Try and feel into the pain. Feel it all the way, so much so that you cant feel the pain. It was bizarre, but it worked. And changed my life. Its a very Buddhist way of leaning into reality, and Ive found it enormously helpful when I feel like my whole body is made of hurt fingers.
2) Take Sensory Care. The normal defenses a well person has against visual, auditory, olfactory, and other kinds of clutter are pretty defeated when youre sick. Your energies are consumed with getting better, not filtering out. This makes it all the more important to be in a tidy, clean space that smells pleasant (or at least clean), sounds peaceful, and looks nice. Even if you cant control all of those things, do what you can with your immediate little sick nesttoss the tissues, make the bed (even if youre in it), put the dishes away, wipe the table down, etc.
3) Ask Yourself: Does This Have Good Energy? Its not about a value judgment, but it usually works out that some things bring us up and other things bring us down. Years ago I came back from months at a yoga retreat and ended up giving away half my booksI looked at each one and asked myself, Positive or negative? When youre sick, your energy is already low, and you dont have energy to waste on artificially cheering up. So make sure that the DVDs, TV shows, books, magazines, people, and anything else you encounter brings you up more than down. For example, no CSI: Special Victims Unit marathons until youre better (or maybe never?).
4) Eat Clean Food. In addition to boosting your recovery with nutrition, eating well while youre sick will ensure you dont have the added food guilt mood baggage of pigging out while not exercising. In other words, go for the comfortsoup, pasta, cerealbut not the bacchanal: Save the Haagen Dazs pint and Krispy Kremes for another day.
5) Drink Plenty of Fresh Water. It will not only hydrate you and flush what needs flushing and keep your cells in optimal fighting shape, it will help your brain counteract negativity. Our moods are very dependent on proper hydration and its easy to forget to drink when youre in nap-and-mope mode. But its physically essential and emotionally nurturing. When I was in chemo they had me drinking obscene amounts of water and it not only kept me less toxic but helped me feel more in the flow and connected to a natural, healing element.
6) Look at Beautiful Things. Just yesterday I was in the parkmy first emergence after the latest cold/flu/fever thingand the first cherry blossoms were blasting off their whites and pinks. It really hit me how soft flowers make me feel inside. Theyre like a soothing visual massage, so gentle when we allow their beauty to permeate our being. Make sure you have a couple of beautiful things nearby when youre illbe it a bouquet of fresh flowers, your fluffy kitty, or a picture of a gorgeous place that makes you happy.
7) Bathe Daily. The dirty (har) secret of sick days is that most people think, Well, Im sick, Im not going anywhere, no need to bathe. But even if youre just going to roll back into bed, first roll into the shower. Use your favorite soaps and potions, visualize the water rinsing the sick off of you. The skins cells actually release little endorphins upon contact with warm liquid. Let those give you a boost, plus youll come out feeling much more shiny and newif still snuffly and achy.
8) Change Your Clothes Every Day, and Make Em Pretty. Again, even if its just your sheets looking at you all day, youll feel 100 times better in a fresh pair of jammies. Ive also recently either mended or tossed my loungewearno matter how belovedwith rips, stains, and other signs of run down disrepair. Its amazing how I can identify less as a sick person when Im in clean, attractive clothes, and more as a person getting well or just relaxing. A good trick for the brain.
9) Dont Isolate. Its especially easy for those who live alone to disconnect from the outside world when theyre sick. Do your best not to let that happen. Call or email friends and let them know youre not feeling well; theyll check in on you. Open to receiving their love while youre at itdont automatically turn down offers of soup and juice and company. Let yourself be nurtured, it will help along the healing process.
10) Laugh. Ok, its the most annoying part of The Secret movie and book to me, but theres a grain of truth in it. A woman says shes healed her breast cancer by watching funny movies.And though I think thats a wildly dangerous and irresponsible thing to say to a mass audience, laughter does release happy chemicals into our bloodstream, it uplifts our spirits, it gives us a reason to live, something to look forward to. So much of getting well is about imbibing joy into our minds and our cells. Our bodies may or may not take the hint, but it certainly cant hurt to give it something to work with.
"Remember.Research is the key to your cure!"
DNA testing for Porphyria
Saturday - January 2, 2016 @ 10:30:11
DNA testing for Porphyria
The Mount Sinai Genetic Testing Laboratory in New York City is proud to announce availability of DNA testing forseven porphyrias, including Acute intermittent Porphyria (AIP), Hereditary Coproporphyria (HCP), Variegate Porphyria (VP), familial Porphyria Cutanea Tarda (f-PCT), Hepatoerythropoietic Porphyria (HEP), Erythropoietic Protoporphyria (EPP) and Congenital Erythropoietc Porphyria (CEP). This is the only laboratory in the United States that offers DNA testing for all of these porphyrias. The testing program was developed with a grant from the American Porphyria Foundation. We thank the Porphyria patients who sent us their blood to develop and validate these tests.
Before requesting DNA testing, it is recommended that patients have biochemical testing (urinary, stool and/or plasma porphyrins and porphyrin precursors (ALA and PBG) and/or enzyme assays). However, many patients have not had an acute attack or are not symptomatic at present, so biochemical testing may be inconclusive.
In contrast, DNA testing is the most accurate and reliable method for determining if a person has a specific porphyria and is considered the "gold standard" for the diagnosis of genetic disorders. If a mutation (or change) in the DNA sequence is found in a specific Porphyria-causing gene, the diagnosis of that Porphyria is confirmed. DNA analysis will detect more than 97% of known disease-causing mutations. DNA testing can be performed whether the patient is symptomatic or not and requires only a small amount of blood sent to the laboratory at room temperature. Once a mutation has been identified, DNA analysis can then be performed on other family members to determine if they have inherited that Porphyria, thus allowing identification of individuals who can be counseled about appropriate management in order to avoid or minimize disease complications.
It is important for patients to realize the limitations of DNA testing. Each porphyria is caused by a mutation in the DNA sequence of a specific gene. Thus, the diagnosis of a specific porphyria determines what gene to test. Diagnosis of the specific porphyria can be difficult because the three acute porphyrias (AIP, HCP, VP) typically have similar acute symptoms, biochemical findings, and responses to treatment. This means, for example, that if a patient has been given the diagnosis of AIP and no AIP gene mutation is identified, it is possible that the patient has a different acute Porphyria. For patients with symptoms of an acute porphyria, but without a specific diagnosis, we offer a "triple test," which includes DNA testing for the three major acute porphyrias (AIP, HCP, and VP).
DNA testing involves sophisticated DNA sequencing which is multi-procedural, labor intensive, and expensive. In the porphyrias, there are no common mutations so the entire gene must be sequenced in each new family.
The cost for DNA testing for one specific porphyria is $802 to $922, depending on the specific porphyria being tested. Once a mutation is identified, the cost for DNA testing of other family members is $226 per person. For patients with symptoms of an acute porphyria, but whose specific porphyria has not been identified, the Acute Porphyrias Panel for analysis of the genes causing the three acute porphyrias can be performed for $1882, a savings of $700 over sequencing each gene separately. Results from DNA testing are typically available in 2 to 4 weeks.
The Mount Sinai Genetic Testing Laboratory, Porphyria DNA Testing Laboratory accepts some insurance; please contact the genetic counselor at the laboratory for current information. Alternatively, payment can be made by credit card or check, which must accompany the patient's blood sample. A receipt will be provided, but depending on the patient's insurance company and policy, the reimbursement will vary and some companies will not pay for Porphyria testing at all. By New York State regulation, DNA testing for any genetic disorder must be ordered by a physician who also must sign the requisition form and who will receive the results. A patient must also read and sign a consent form which explains DNA testing and offers counseling.
Please contact the genetic counselor, Dana Doheny, MS, CGC, by email (porphyria@mssm.edu or dana.doheny@mssm.edu) or telephone (212-659-6779, direct-line; 866-322-7963, toll-free) for the required forms (including sample requirements, shipping instructions, test requisition, and consent form), to arrange for testing, and/or for additional information.
The Mount Sinai Genetic Testing Laboratory, Porphyria DNA Testing Laboratory, Department of Genetics & Genomic Sciences, The Mount Sinai School of Medicine, 1425 Madison Avenue, Room 14-74, Box 1498, New York City, NY 10029-6574.
Sorry you went through so much but glad some one w
Wednesday - September 28, 2016 @ 10:34:13
Sorry you went through so much but glad some one was able to figure it out for you. I know what you mean by trying to figure out if the pain will be an attack or just the pain for the day.
Although it may sound a little morbid I am happy t
Wednesday - October 5, 2016 @ 07:32:25
Although it may sound a little morbid I am happy to read about someone else who has went through the same agony, psychologically and physically, of everyone telling you that you're fine while you wither in pain. It took me 6 years until I was diagnosed with a porphyria and I am still fighting my doctor to do further testing to verify which one. I think I have PCT but until I get tested for ALA and PBG I cannot be officially diagnosed and until I am officially diagnosed I cannot receive medication or treatment. But the doctors just refuse to help. I recently contacted the APF and they are sending information to my doctor so that he understands what I am going through. My first symptoms were a surprise seizure on Christmas eve 2006, then nothing until 4 years later I started getting these blisters from going out in the sun, then doctor after doctor; dermatologists, endocrinologists, urologists, ENTs, several different primary care physicians all of which just didn't know what to do and either said its all in my head or they prescribed me with topical cortisone creams (for the blisters) and anti anxiety meds. Then one day I went to a gastroenterologist for suspected Crohn's and he just happened to test my urine for porphryins (along with the other gallon of blood he got removed for testing and low and behold there it was. Even with that urine test and having it retested my now hematologist is still reluctant to do the final and recommended tests.
The biggest obstacle that I have been facing is that each doctor just shrugs their shoulders and moves on with their life, not even recommending me to any sort of specialist. I had to do the research myself and contact almost all of these specialists myself, some of which were not covered under my insurance and I have been paying out of pocket. If it were not for my own research and action I may have never been diagnosed. Doing your own research seems to be a common theme amongst people with porphyria due to its rarity and I cannot stress this enough how important it is. Once I found out I had porphyria I have not touched a drop of alcohol and I haven't had an incident in 6 weeks that's the longest I have gone without incident in 6 years (previous record was 2.5 weeks). Thanks for sharing Lina.
Wow, you've been through so much, your story i
Saturday - October 29, 2016 @ 03:18:46
Wow, you've been through so much, your story is so similar to mine, after highschool, I took a year off from school to help my mom out at home, when I was working at 21 yrs old a few years later, I had always had symptoms of porphyria all my life, but I developed my first really severe attack that day at 21 and started throwing up right after eating, I couldn't even keep liquid down and would have straight up dry heaves and throw up pure bile, I went to the ER and they attributed all my symptoms, abdominal pain, vommitting- to an ovarian cyst, they recommended me for emergency surgery to remove it to relieve my symptoms of pain and nausea/sickness, but not one of us could have known I was in an aip attack and needed heme and IV glucose only. I desperately agreed to have the surgery, trusting in my doctors and I did actually have an ovarian cyst, many women have them, they are a common problem and funny enough commmon with porphyria in women I've found later on- anyway, I agreed to have the surgery because I was so sick and desperate for some kind of relief and the next morning I woke up from surgery, it looked there was like blood in my urine, blood red, freaked my nurse out and I was a little concerned but I tried to stay calm and assume it was from surgery, luckily it went away quickly, but then my Blood pressure went sky high and I mean dangerously high- I'd all my life had low blood pressure, this was the first time I remember the words high blood pressure being used associated with me, my pain levels, which were really bad intolerable already, tripled- it felt like someone was going over my entire body with a straight razor, scraping the skin. I started moaning in pain and scared the other sick ppl in my hospital room. I developed a severely bad reaction to the surgery site, they said I'd had an allergic reaction to the Co2 used to expand my stomach during surgery, I was extremely thin at the time, not being able to keep food down. So it felt like nails were scraping me, burned like fire, ontop of the abdominal pain from the increased attack. They sent me home in really worse shape than when I'd arrived and after going back to the ER again after my surgery, desperate for some answers and relief, they diagnosed me with the flu and sent me home again, unable to find anything abnormal they said. That night I felt like I was literally dying. So the next morning I had had not much rest and I get up to use the restroom, I'm coming out and my parents said I bent down to move a fan and was very irritable, the next thing I know I'm waking up on the bathroom floor with my StepDad holding me down. I had collapsed in front of my parents and went into full gran mal seizure in front of them on the floor, I had never had a seizure in my life before that moment. I stopped breathing they told me, but I came back awake, but was very disoriented and irritable, I struggled with my stepdad, tearing at his shirt a couple times, they said I did that a couple times- once at home and once in the hospital later on when I collapsed again and went into like a catatonic state for a couple minutes, my sodium was bottomed out- it freaked my doctors out. Just uncontrollable frustration that would control me until the pressure built to painful levels and I had to yell or do something to relieve it, it's quite indescribeable, it's like you come back to yourself after it happens and you can never imagine doing anything like that. I'm so happy that you got your heme treatments and got better, I suffered from a prolonged untreated attack and still have severe nerve damage and need a wheelchair, but 3 heme treatments I feel saved my life and I haven't had such a severe attack since then, we know what to do now more for them and prevent them- so nore seizures for me yay, although I still get petit mals, spasms at times in my legs and body. Blessings
The porphyria is a group of various ailments. The
Wednesday - November 9, 2016 @ 12:51:32
The porphyria is a group of various ailments. The basic Porphyria Causes is an imperfect chemical essential to the heme biosynthesis pathway. Porphyria Natural Treatment that together with the particular treatment can work ponders for everybody.
Is this a study where patients will receive Scenes
Monday - December 5, 2016 @ 18:07:21
Is this a study where patients will receive Scenesse ?
Where can I get brochures on AIP? I want to hand t
Tuesday - December 27, 2016 @ 00:41:14
Where can I get brochures on AIP? I want to hand them out to everyone in the hospital when my daughter has an attack. They are fairly frequent and we think stress is the main correlation between attacks. Have you heard of stress and porphyria?









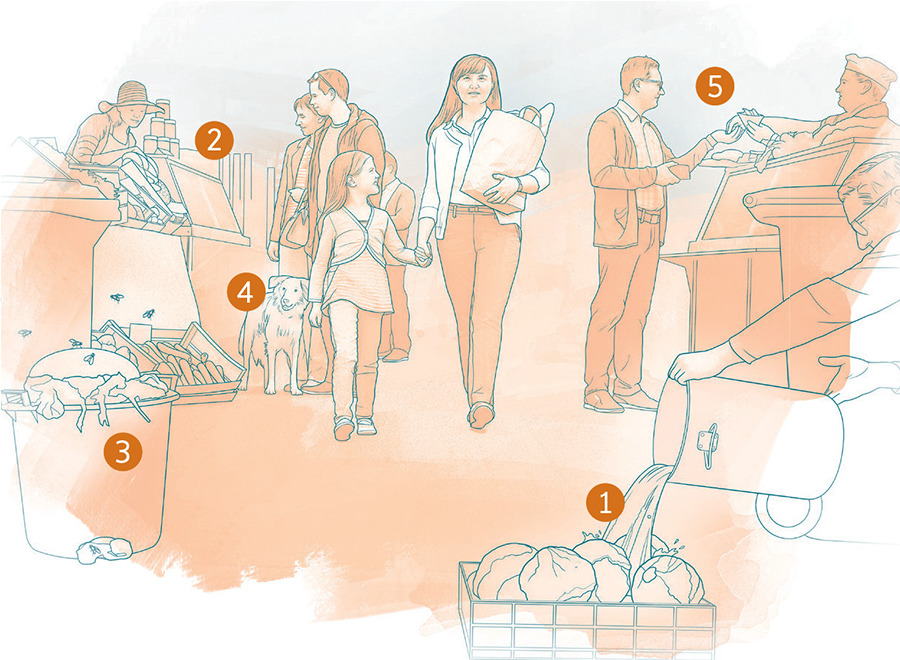
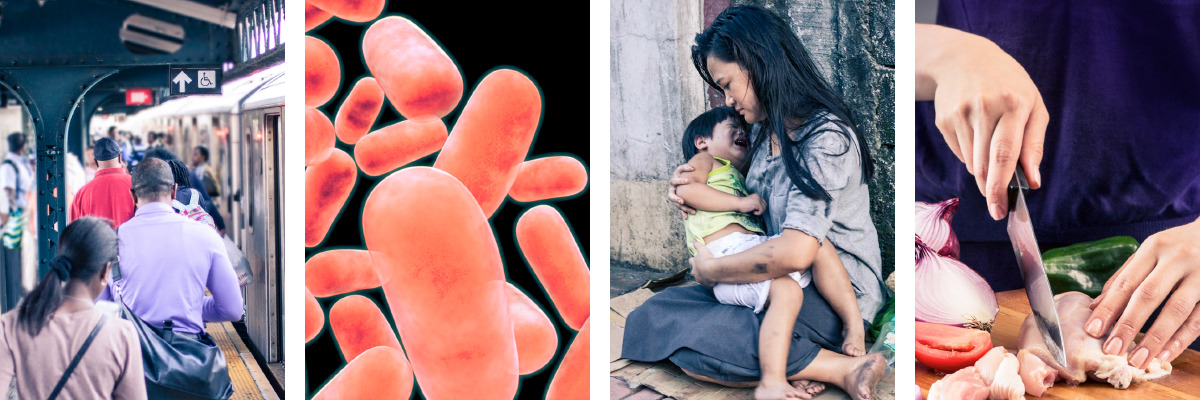




















































 I am a 38-year-old mother of two who lives right outside of Philadelphia. I was diagnosed with EPP at the age of two. My mother had noticed that I would get scabs on my nose when she took me out in the sun, so she took me to our family doctor who thought I had porphyria. Since this wasn't something anyone in our family had she got a second opinion at Children's Hospital of Philadelphia where they confirmed, through testing, that I did indeed have Erythropoietic Protoporphyria (EPP).
I am a 38-year-old mother of two who lives right outside of Philadelphia. I was diagnosed with EPP at the age of two. My mother had noticed that I would get scabs on my nose when she took me out in the sun, so she took me to our family doctor who thought I had porphyria. Since this wasn't something anyone in our family had she got a second opinion at Children's Hospital of Philadelphia where they confirmed, through testing, that I did indeed have Erythropoietic Protoporphyria (EPP). decided that we should test the baby right after birth to determine if he/she would have EPP. When my daughter was born we had her tested and she tested positive for it. When my son was born, he too tested positive for it. The doctors recommended that we retest them after they were two, as the tests they had as newborns could be a false positive.
decided that we should test the baby right after birth to determine if he/she would have EPP. When my daughter was born we had her tested and she tested positive for it. When my son was born, he too tested positive for it. The doctors recommended that we retest them after they were two, as the tests they had as newborns could be a false positive. I try to stay out of the sun as much as possible but things like sun exposure through the car windows when I am driving can aggravate EPP. I now use gloves while driving in the spring and summer and a sun-protective scarf when I am a passenger. When I was younger I thought the hardest part was missing out on all the fun stuff myself, but after having kids I find that they have had to adapt to my limitations too and at times that makes me very sad. When they were babies and I had an outbreak, changing a diaper was a huge and very painful task and holding them was no better.
I try to stay out of the sun as much as possible but things like sun exposure through the car windows when I am driving can aggravate EPP. I now use gloves while driving in the spring and summer and a sun-protective scarf when I am a passenger. When I was younger I thought the hardest part was missing out on all the fun stuff myself, but after having kids I find that they have had to adapt to my limitations too and at times that makes me very sad. When they were babies and I had an outbreak, changing a diaper was a huge and very painful task and holding them was no better.








 Having Children
Having Children

















![[Picture on page 3]](https://assetsnffrgf-a.akamaihd.net/assets/m/g11/201103/g11_201103_E/102011081_univ_cnt_1_xl.jpg)






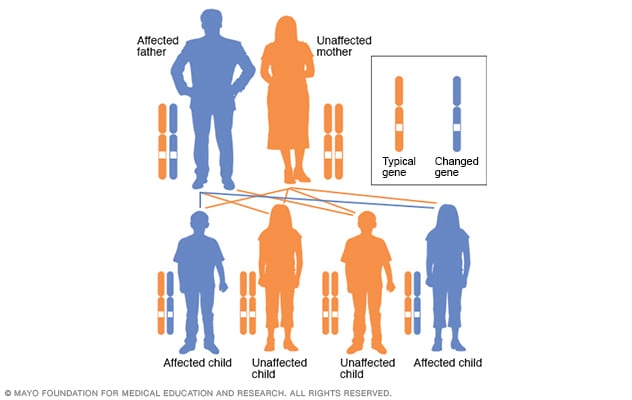
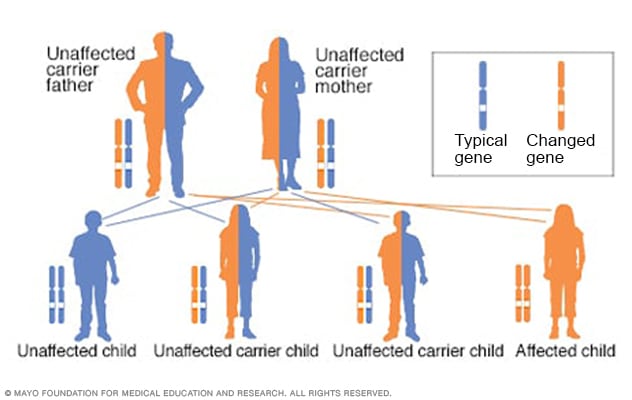
 The Mount Sinai Genetic Testing Laboratory in New York City is proud to announce availability of DNA testing forseven porphyrias, including Acute intermittent Porphyria (AIP), Hereditary Coproporphyria (HCP), Variegate Porphyria (VP), familial Porphyria Cutanea Tarda (f-PCT), Hepatoerythropoietic Porphyria (HEP), Erythropoietic Protoporphyria (EPP) and Congenital Erythropoietc Porphyria (CEP). This is the only laboratory in the United States that offers DNA testing for all of these porphyrias. The testing program was developed with a grant from the American Porphyria Foundation. We thank the Porphyria patients who sent us their blood to develop and validate these tests.
The Mount Sinai Genetic Testing Laboratory in New York City is proud to announce availability of DNA testing forseven porphyrias, including Acute intermittent Porphyria (AIP), Hereditary Coproporphyria (HCP), Variegate Porphyria (VP), familial Porphyria Cutanea Tarda (f-PCT), Hepatoerythropoietic Porphyria (HEP), Erythropoietic Protoporphyria (EPP) and Congenital Erythropoietc Porphyria (CEP). This is the only laboratory in the United States that offers DNA testing for all of these porphyrias. The testing program was developed with a grant from the American Porphyria Foundation. We thank the Porphyria patients who sent us their blood to develop and validate these tests.{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}