Have you ever been involved in Clinical Studies or an observational study? Please give serious thought to joining. Please contact the APF 1.866.APF.3635
Why Join Research?
There are two main types of clinical studies: clinical trials and observational studies. In a clinical trial, there is some form of treatment intervention. There is no intervention in an observational study, which is aimed at observing patients to better understand the long-term course of their disease.
Clinical trials are used to test new treatments before they are approved for use by the FDA. This type of trial gives patients a chance to try out a new medication in its early stages. As with any experiment, the result of a trial is not known before its conclusion. Your participation could help demonstrate a terrific treatment breakthrough, or it could help scientists discover that a new treatment does not work after all. There may be some risk involved from the treatment in a clinical trial.
Participating in either a clinical trial or an observational study is a serious responsibility. Volunteering to participate could be a way to help yourself, affected family members and other patients by advancing medical and scientific knowledge of your condition. Some patients derive great satisfaction from assisting doctors in the study of their disease. Participation in a study can also mean a chance to meet a porphyria researcher in a clinical setting, and the consultation can be beneficial.
For information on porphyria trials currently recruiting patients, call the APF office or visit NIH’s clinical trials website: “Clinical Trials of Medical Treatments: Why Volunteer?” might also be useful reading as you think about whether you’d like to participate or not.
How rare is RARE
Monday - December 23, 2019 @ 06:30:00
A disease or disorder is defined as rare in Europe when it affects fewer than 1 in 2000. A disease or disorder is defined as rare in the USA when it affects fewer than 200,000 Americans at any given time. ... In the EU, as many as 30 million people may be affected by one of over 6000 existing rare diseases. Porphyria is just one of those ultra rare diseases in the form of Acute Hepatic Porphyria or Cutaneous type of Porphyria. If you or a loved one suffers from this debilitating disease and need help please contact the American Porphyria Foundation @ 1.866.APF.3635 and you can learn form Experts Physicians from around the world. porphyriafoundation.org
Informative Porphyria Post
Thursday - December 19, 2019 @ 06:30:00
Now Available on the APF Website - the 4th Quarter Newsletter
The 4th Quarter APF newsletter has been mailed and is hitting doorsteps as we speak. In case you missed the latest copy, you can read it on the APF website.
Our very own APF store is your perfect solution for last minute gifts and stocking stuffers.
The store has items to fit everyone's needs. There, you can find cozy tee-shirts, fun apparel Items - not to mention practical items everyone should have. By shopping at the APF Store, you can spread the holiday spirit, support a great cause and give to those you love.
Its almost a wrap! Remember, when you finish your holiday shopping at smile.amazon.com, amazonsmile donates to American Porphyria Foundation at no cost to you!
You can also shop all year round using amazonsmile and the donations will still go to the APF.
It's simple.You shop. Amazon gives.
Amazon donates 0.5% of the price of your eligible amazonsmile purchases to the APF.
amazonsmile is the same Amazon you know. Same products, same prices, same service. It's simple and easy!
Use the amazonsmile Link for Your Holiday Shopping
Amazon store
During this holiday season, we would like to wish all of you good cheer and health. May your holidays be blessed with love and caring. We look forward to a New Year of continued advancements in research, education and patient advocacy.
APF Holiday Office Hours
In honor of the holiday season our office will be closed from Wednesday December 25, 2019 through Wednesday January 1, 2019.
Should you experience a medical emergency and need assistance, please call 911 immediately.
Acute intermittent porphyria mimics a variety of commonly occurring disorders and thus poses a diagnostic quagmire. Psychiatric manifestations include hysteria, anxiety, depression, phobias, psychosis, organic disorders, agitation, delirium, and altered consciousness ranging from somnolence to coma.
About the Porphyria's. Have you looked to see all the types there are? Which one have you been diagnosed with? What type of therapy or treatment is available for you? Many of you are new to Porphyria and are trying to get a proper diagnosis. This information comes directly from the APF. If you need help in getting a diagnosis or have a questions on the different types please give the APF a call 1.866.APF.3635. www.porphyriafoundation.org
About Porphyria
Porphyria is not a single disease but a group of eight inherited genetic disorders that differ considerably from each other. A common feature in all Porphyrias is the accumulation in the body of porphyrins or porphyrin precursors. Although these are normal body chemicals, they normally do not accumulate. Precisely which of these chemicals builds up depends on the type of Porphyria.
The terms porphyrin and porphyria are derived from the Greek word porphyrus, meaning purple. Urine from some Porphyria patients may be reddish-purple in color due to the presence of excess porphyrins and related substances in the urine, and the urine may darken after exposure to light.
Cause
Porphyria arises as a result of a malfunction in one of the eight steps in the body's synthesis of a complex molecule called heme. Heme is essential for the transport of oxygen to cells in the body. If any step in the synthesis of heme is blocked, an intermediate chemical accumulates in the cell, resulting in oxygen depletion. Those intermediate chemicals, known as porphyrins or porphyrin precursors, are the substances of which heme is composed. Each type of Porphyria represents a deficiency of a specific enzyme needed for the synthesis of heme.
Most commonly the Porphyrias are divided into the “acute” and “cutaneous” Porphyrias, depending on the primary symptoms. For many with one of the four acute Porphyrias, Porphyria attacks generally evolve and become more severe over several days, especially the abdominal pain; two of these, Variegate Porphyria and Hereditary Coproporphyria, may also have skin symptoms of blistering after sun exposure. The cutaneous Porphyrias present with blistering and scarring of the skin, pain, and/or redness and swelling in sun-exposed areas.
The Acute Porphyrias
There are four types of acute Porphyrias: Acute Intermittent Porphyria (AIP), Hereditary Coproporphyria (HCP), Variegate Porphyria (VP), and d-aminolevulinic acid dehydratase (ALAD) Porphyria (ADP), which are characterized by episodes of debilitating attacks. These are genetic disorders that are very rare and may be difficult to diagnose for this reason. It is estimated that about 1 in 10,000 Europeans or people of European ancestry have a mutation in one of the genes that cause AIP, VP or HCP. These mutations have been found in all races and many other ethnicities in addition to Europeans.
Approximately 80-90% of individuals who carry a gene mutation for Acute Intermittent Porphyria, Variegate Porphyria, and Hereditary Coproporphyria remain asymptomatic, and others may have only one or a few acute attacks throughout life. The most frequent symptom is severe abdominal pain and is often accompanied by nausea, vomiting, and constipation. Other symptoms may include heart palpitations, seizures, and hallucinations. People with VP and HCP may also have skin symptoms of blistering after sun exposure.
The Cutaneous Porphyrias
All but one of the cutaneous Porphyrias cause skin blistering and fragility on sun-exposed areas of the body, most commonly the backs of the hands, forearms, face, ears and neck. The cutaneous Porphyrias are: Porphyria Cutanea Tarda (PCT), Hepatoerythropoietic Porphyria (HEP), Congenital Erythropoietic Porphyria (CEP), Erythropoietic Protoporphyria (EPP), and X-linked Protoporphyria (XLP). CEP and HEP occur in childhood with severe blistering skin lesions. PCT occurs in adulthood generally and has less severe blistering skin lesions after sun exposure. Erythropoietic Protoporphyria (EPP) and X-linked Protoporphyria (XLP) have the same symptoms of painful, but non-blistering, reactions to sunlight. There can also be swelling and redness of the sun exposed areas of the skin with EPP and XLP.
Prevalence
The Porphyrias are rare diseases. Taken together, all forms of Porphyria afflict fewer than 200,000 people in the United States. Based on European studies, the prevalence of the most common Porphyria, Porphyria Cutanea Tarda (PCT), is 1 in 10,000; the most common acute Porphyria, Acute Intermittent Porphyria (AlP), is about 1 in 20,000; and the most common erythropoietic Porphyria, Erythropoietic Protoporphyria (EPP), is estimated at 1 in 50,000 to 75,000. Congenital Erythropoietic Porphyria (CEP) is extremely rare with prevalence estimates of 1 in 1,000,000 or less. Only 9 cases of ALAD-deficiency Porphyria (ADP) are documented.
Signs and Symptoms
The signs and symptoms and vary significantly from one type of Porphyria to the next. Because the symptoms of the various Porphyrias may resemble symptoms of other more common disorders, diagnosis may be difficult. The onset, severity and type of symptoms can vary greatly in individuals with a specific type of Porphyria. This variation may depend on, in part, the amount of residual enzyme activity in each individual. Individuals with more significant enzyme deficiency may have more severe symptoms and earlier onset. Individuals with partial deficiency will have milder symptoms, and some individuals will not develop any symptoms (asymptomatic). It is important to note that affected individuals may not have all of the symptoms discussed below. Affected individuals should talk to their medical team about their specific case, associated symptoms and overall prognosis.
Acute Intermittent Porphyria
Abdominal pain is the most common complaint in Acute Intermittent Porphyria (AIP). In addition, some of the following symptoms occur with varying frequency: pain in the arms and leg, generalized weakness, vomiting, confusion, constipation, tachycardia, fluctuating blood pressure, urinary retention, psychosis, hallucinations, and seizures. The muscle weakness may progress to respiratory paralysis, necessitating artificial respiration. Porphobilinogen (PBG) and Aminolevulenic Acid (ALA), porphyrin precursors, are elevated during the attack but may be consistently high in some patients. Urine may exhibit a purple-red color. Unlike other forms of Porphyria, sun sensitivity is not present in this type.
Variegate Porphyria
Variegate Porphyria (VP) is characterized by abrasions, blisters, and erosions of the skin which are commonly seen during the second and third decade. These lesions tend to heal slowly, often leaving pigmented or slightly depressed scars. The patients experience sensitivity to light and fragility of skin exposed to the sun. Although in many patients’ manifestations remain limited to the skin, episodes similar to those of acute Porphyria are not uncommon. Therefore, the symptoms, drugs, precautionary measures, and treatment described for Acute Intermittent Porphyria are applicable to Variegate Porphyria.
Hereditary Coproporphyria
The large amounts of coproporphyrin present in Hereditary Coproporphyria (HCP) makes the patient sensitive to sunlight, but skin disease is rarely severe in this type of Porphyria. Clinically it resembles Variegate Porphyria except that a larger percentage of affected individuals exhibit few symptoms. Other symptoms are similar to those listed for Acute Intermittent Porphyria.
Erythropoietic Protoporphyria
Erythropoietic Protoporphyria (EPP) can have mild to severe light sensitivity and burning on exposure to the sunlight. Usually, the symptoms subside in twelve to twenty-four hours and heal without significant scarring or discoloration to the skin. The skin lesions may also progress to a chronic stage persisting for weeks and healing with a superficial scar. These manifestations generally begin in childhood. They are more severe in the summer and can recur throughout life. Affected skin, at times, exhibits the fragility or blister formation seen in other photosensitizing types of Porphyria. Liver dysfunction along with gallbladder, bile duct or bile involvement (hepatobiliary system) may be associated with significant liver damage.
Porphyria Cutanea Tarda
In Porphyria Cutanea Tarda (PCT), exposed skin shows abnormalities similar to those found in Variegate Porphyria. They range from slight fragility of the skin to persistent scarring and disfiguration. Due to fragility of the skin, minor trauma may induce blister formation. Areas of increased and decreased pigment content may be noted on the skin. Blistering of light exposed skin and increased hair growth, especially on the face, are also characteristic.
Congenital Erythropoietic Porphyria
Congenital Erythropoietic Porphyria (CEP) is a very rare disease with approximately 150 patients reported in the world. CEP is often manifested shortly after birth with dark urine and sunlight sensitivity causing blistering and skin fragility. Later, brownish, fluorescent teeth, increased hair growth, and pronounced scarring may occur. In some cases, loss of fingers and toes and cartilage from ears or nose may be noted.
ALA-D Porphyria
ALA-D Porphyria (ADP) symptoms usually arise from effects on the nervous system and/or the skin. Sometimes, the cause of the nervous system symptoms is not clear. Skin manifestations include burning, blistering, and scarring of the sun exposed areas. The disease usually manifests after puberty, and more commonly occurs in women. The most common symptom is severe abdominal pain. Other characteristics are nausea, vomiting, rapid heart rate, increased blood pressure, confusion and/or seizures, and the passing of ALA (delta-aminolevulinic acid) in the urine.
LIVE FB WORLDWIDE JOIN US
Tuesday - December 3, 2019 @ 06:30:00
Join Porphyria American Porphyria Foundation to listen to Dr. Bruce Wang and ask your questions!
Porphyria Members: Dont forget to set your alarm to 6:45pm
What would you ask a porphyria expert if they were sitting in front of you??
Youll have that opportunity on December 4, 7-8pm EST and December 5, 1-2pm EST!
Join Porphyria American Porphyria Foundation to listen to Dr. Bruce Wang and ask your questions!
Today is #GivingTuesday! Help us today by donating now using the link below and sharing it with friends. Your contribution will make an impact, whether you donate $5 or $500. Every little bit helps.
GivingTuesday is a global day of giving
celebrated the Tuesday after Thanksgiving in the United States. Please consider
supporting the APF on Tuesday, December 3rd.
When you support the APF, you are making a
contribution to an organization that provides solid porphyria education, hope
and essential support to those affected by porphyria. You can be proud to be an
integral part of the help we provide patients on the road to accurate diagnosis,
proper care, and hopefully someday a cure. This year our goal is set for
5000.00$
{The APF has 3,262 FB Members if each FB member donated 1.54 $ each we would make
our GOAL!!!}
Donations will be matched dollar for dollar on a first-come, first-served basis.
Matching is available
for any eligible US-based 501(c)(3) nonprofit that can receive donations on
Facebook. Donations up to $250k per nonprofit and $20k per donor are eligible
to be matched. We invite you to give Tuesday.
To Donate to the APF on Giving Tuesday Please Click Here
Dr. Wang is going LIVE in 5 days! Join the Porphyria- American Porphyria Foundation
Friday - November 29, 2019 @ 06:30:00
Dr. Wang is going LIVE in 5 days!
Join the Porphyria- American Porphyria Foundation
Head there now so you will be all set to listen in and ask your questions for December 4, 7-8pm EST and December 5, 1-2pm EST!
Happy Thanksgiving!
Thursday - November 28, 2019 @ 06:30:00
Happy Thanksgiving!
We are thankful that you all are part of the APF Family and our warmest wishes for a happy holiday go out to you and yours.
The APF Office will be closed Thursday and Friday, November 28 & 29, 2019 for the Thanksgiving Holiday.
Porphyria Post
Wednesday - November 27, 2019 @ 06:30:00
Facebook Live with Dr. Bruce Wang
MARK YOUR CALENDARS!
?Wednesday, December 4, 7-8 pm EST
and
Thursday, December 5, 1-2 pm EST
The APF will be holding two Question and Answer Facebook Live sessions with Porphyria expert Dr. Bruce Wang, of the University of California, San Francisco Porphyria Center.
We are very excited and appreciative of Dr. Wang for sharing his expertise with our community. These will be exceptional sessions that you won't want to miss!
To Join Dr. Wang's Session Click on our Facebook Link
GivingTuesday is a global day of giving celebrated the Tuesday after Thanksgiving in the United States.
Please consider supporting the APF on Tuesday, December 3rd.
When you support the APF, you are making a contribution to an organization that provides solid porphyria education, hope and essential support to those affected by porphyria. You can be proud to be an integral part of the help we provide patients on the road to accurate diagnosis, proper care, and hopefully someday a cure.
To Donate to the APF on Giving Tuesday Please Click Here
Happy Thanksgiving!
We are thankful that you all are part of the APF Family and our warmest wishes for a happy holiday go out to you and yours.
The APF Office will be closed Thursday and Friday, November 28 & 29, 2019 for the Thanksgiving Holiday.
Facebook Live with Dr. Bruce Wang
Wednesday - November 27, 2019 @ 06:30:00
By
Purple Light
November 27, 2019
Facebook Live with Dr. Bruce Wang MARK YOUR CALENDARS! Wednesday, December 4, 7-8 pm EST & Thursday, December 5, 1-2 pm EST
The APF will be holding two Question and Answer Facebook Live sessions with Porphyria expert Dr. Bruce Wang, of the University of California, San Francisco Porphyria Center.
We are very excited and appreciative of Dr. Wang for sharing his expertise with our community. These will be exceptional sessions that you won't want to miss!
The American Porphyria Foundation is pleased to announce that the
Food and Drug Administration has granted APPROVAL for GIVLAARI (Givosiran) for
the treatment of Acute Hepatic Porphyria!
US patients will now have access to this preventative RNAi
treatment. This approval is the first step in gaining access to GIVLAARI
(Givosiran) in the US. Availability to patients will progress as Alnylam
Pharmaceuticals pivots from FDA approval to commercial availability.
The APF will update you frequently on progress related to access
and distribution of GIVLAARI (Givosiran) treatment.
Congratulations to YOU, our patient community, who participated in
the pivotal Phase 2 and Phase 3 clinical trials that led to this approval. You
are our medical heroes!
The Protect the Future campaign was established to attract and train the next generation of doctors and specialists in the field of Porphyria. Over the next decade, we will lose ninety percent of our valued Porphyria experts. These men and women have led Porphyria research, testing and treatment for the past 30 years. Without financial support, we run the risk of losing knowledge of the disease, quality testing, diagnosis, and treatment, and ultimately a cure.
Protect the Future supports young doctors who work and study with long-time experts, seeing patients and doing research, in order to gain the expertise they will need to care for the U.S. Porphyria patient population for decades to come.
Protect the Future is a separate fundraising campaign with its own specific needs. We are seeking additional donations for the specific purpose of locating and training new Porphyria specialists.
You and I are pulled in so many directions for our donations, such as churches, police leagues and schools. But rarely will we be presented with an opportunity to affect the course of our own lives and those of our children for years to come.
Let’s work together to bring another great specialist into our lives. We are so very fortunate to have the few Porphyria experts who are presently in the field. Now we can protect our own future by adding more doctors.
Donate now! Please make a note that you are donating to the Protect the Future campaign so that we can allocate those funds appropriately.
undiagnosed condition?
Tuesday - November 12, 2019 @ 06:30:00
Trying to find an underlying diagnosis for many conditions can be a very long and frustrating experience. With more rare conditions, a diagnosis can often take many years. Although this can be incredibly difficult, the following information may help you navigate through the process of trying to obtain a diagnosis. If YOU suspect you have one of the Porphyrias please contact the American Porphyria Foundation 1.866.APF.3635 or visit porphyriafoundation.org
Are there research programs available for people without a diagnosis? Yes. If an individual’s health care providers and specialists have not been able to make a definite diagnosis so far, participating in a research study or clinical trial may be another option. See below for a description of some of the National Institutes of Health (NIH) research programs that are going on now:
The Undiagnosed Diseases Network (UDN) is a research study funded by the National Institutes of Health Common Fund. The UDN is made up of clinical and research centers across the United States working to improve diagnosis and care of patients with undiagnosed diseases. Physicians and patients with additional questions may call 1-844-746-4836 (1-844-Ring-UDN).
ClinicalTrials.gov is database that provides current information on clinical research studies. You can search ClinicalTrials.gov for research studies looking at general categories of diseases (e.g. neurological diseases or eye diseases) or specific symptoms. Some studies accept individuals without a diagnosis with the research goal of making a diagnosis.
One study that is enrolling individuals who do not have a diagnosis is entitled "Pediatric patients with Metabolic and Other Genetic Diseases". This study is evaluating individuals with known or suspected genetic diseases, including metabolic diseases. Despite the name, people of all ages may be eligible for this study.
To find out more about clinical trials that take place at the NIH, you can call the NIH Clinical Center to talk to a specialist.
Patient Recruitment and Public Liaison Office NIH Clinical Center National Institutes of Health Bethesda, Maryland 20892-2655 Toll-free: (800) 411-1222 Fax: (301) 480-9793 E-mail: prpl@mail.cc.nih.gov
How can I learn more about clinical trials? If you or someone you know is interested in enrolling in a clinical trial, you can find helpful general information on clinical trials at ClinicalTrials.gov.
Resources on many charitable or special-fare flights to research and treatment sites and low-cost hospitality accommodations for outpatients and family members, as well as ambulance services, are listed in the GARD Help with Travel Costs guide.
Are there any advocacy groups for people with an undiagnosed condition? Yes. See below for additional information and supportive resources for individuals with an undiagnosed condition and their families.
Syndromes Without A Name (SWAN) is a supportive organization for families of children who have undiagnosed, unnamed conditions, or who are still looking for a diagnosis.
Syndromes Without A Name (SWAN) United States Toll-free: 888-880-SWAN Telephone: 269-692-2090 E-mail: info@swanusa.org Web site: http://swanusa.org/
The National Organization for Rare Disorders (NORD) is a federation of more than 130 nonprofit voluntary health organizations serving people with rare disorders. The NORD Web site includes information on medication assistance programs and networking programs, a resource guide, and links to other online resources. You can get this information through NORD's Web site or by calling or writing the NORD offices.
National Organization for Rare Disorders 55 Kenosia Avenue PO Box 1968 Danbury, CT 06813-1968 Toll free: 800-999-6673 (voicemail only) Telephone: 203-744-0100 TDD: 203-797-9590 Fax: 203-798-2291 E-mail: orphan@rarediseases.org Web site: http://www.rarediseases.org/
NORD recognizes that patients and families coping with undiagnosed rare medical conditions may experience unique challenges in accessing appropriate medical care and social support. Undiagnosed patients include those who are “not yet diagnosed” because they have not been referred to the appropriate medical specialist as well as patients who have a condition not previously described and for which a diagnostic test is not yet available.
Who should I talk to if I have financial concerns? It can sometimes take many years of specialized appointments and testing for a condition to be diagnosed, and this affects many individuals and families financially.
The Patient Advocate Foundation is a non-profit organization that serves as a liaison between families and their insurer, employer or creditors to resolve insurance, job retention and/or debt crisis matters related to their medical conditions. You can contact the Patient Advocate Foundation for further information.
Are there organizations that can help with the cost of travel? Yes. Traveling to specialized centers for testing and diagnosis can be costly; the following organizations help organize free travel for patients within the US.
Where can I find out more about financial assistance? The National Organization for Rare Disorders (NORD) provides information on financial and medication assistance programs, health insurance, medicare/medicaid programs, and links to additional online resources. Most of these resources are available only to individuals in the United States.
Mitsubishi Phase 2 Clinical Trial Meets Endpoint for Treatment of Erythropoietic Protoporphyria (EPP)
Mitsubishi Tanabe Pharma Development America, Inc. today announced successful completion of the Phase 2 clinical trial of
MT-7117, an investigational oral treatment under development for the
prevention of phototoxicity (including severe pain on exposure to
sunlight) in patients with Erythropoietic Protoporphyria (EPP).
In this Phase 2 proof of concept trial, MT-7117 met its
primary endpoint and was generally well tolerated with an acceptable
safety profile.
'The results of ENDEAVOR are very encouraging and
will pave the way for a pivotal trial to evaluate the safety, efficacy
and effectiveness of MT-7117 as an oral once a day treatment option for
EPP' said Robert Desnick, MD PhD, Dean for Genetic and Genomic Medicine,
Icahn School of Medicine at Mount Sinai, NY and Lead Investigator on the
study.
'If confirmed and approved by regulatory agencies, this could be the first oral treatment option for EPP and we believe could be a
clinically meaningful alternative for patients.'
All items, prices and sizes are on the website. These include tax & shipping. If you have any questions regarding placing an order please email amy@porphyriafoundation.org
Gift giving is just around the corner show your loved one you support them. Did you know that 100% of your purchase goes right back to the APF?
Have You Joined Our Open & Closed Facebook Groups?
Porphyria Research Group?
Have you checked out the APF Facebook groups recently?
Did you know APF has seven Facebook groups? We moderate one public group along with six private disease-specific groups that you can request to join.
The communities are very active and it is a great way to connect with other porphyria patients and caregivers. In addition, the APF regularly posts publications, research,
updates and other educational information As an ultra-rare condition it can seem like you are alone but YOU ARE NOT! come join us!
Below are the Official 7 APF Facebook Groups
Public Group
Porphyria - American Porphyria Foundation
Private Groups
APF: Porphyria - AIP, HCP, VP
APF: Porphyria - PCT
APF: Porphyria - EPP
APF: Porphria - CEP
Porphyria International Support & Education Group
Porphyria Research
SCENESSE In Australia
Have You Joined Our Open & Closed Facebook Groups?
Porphyria Research Group?
Have you checked out the APF Facebook groups recently?
Did you know APF has seven Facebook groups? We moderate one public group along with six private disease-specific groups that you can request to join.
The communities are very active and it is a great way to connect with other porphyria patients and caregivers. In addition, the APF regularly posts publications, research,
updates and other educational information As an ultra-rare condition it can seem like you are alone but YOU ARE NOT! come join us!
Below are the Official 7 APF Facebook Groups
Public Group
Porphyria - American Porphyria Foundation
Private Groups
APF: Porphyria - AIP, HCP, VP
APF: Porphyria - PCT
APF: Porphyria - EPP
APF: Porphria - CEP
Porphyria International Support & Education Group
Porphyria Research
SCENESSE In Australia
The Australian Therapeutic Goods Administration (TGA) has enabled Priority Registration Pathway for SCENESSE for the treatment of Erythropoietic Protoporphyria (EPP). This review pathway gives a target time frame of 150 working days from submission of the a new drug application.
The APF congratulates the Australian EPP community on this progress! SCENESSE was approved by the US Food and Drug Administration on October 8, 2019. Distribution and reimbursement arrangements are in preparation.
Need Something from the APF?
Were Here for You!
Call us on 866-APF-3635
Click this link to email us at porphyrus@porphyriafoundation.org
Check us out on www.porphyriafoundation.org
RSVP TODAY
Patient Education and Support Meeting - Orlando
LOCATION
Hilton Garden Inn, Poseidon/Neptune Room, Orlando at SeaWorld 6850 Westwood Blvd, Orlando, FL 32821
DATE AND TIME
12/08/19 6:30pm - 12/08/19 8:30pm
The American Porphyria Foundation will be hosting a Patient Education and Support Meeting in Orlando, FL on Sunday, December 8. RSVP TODAY!
I'll be there!
Maybe
I can't make it
Patient Education and Support Meeting - Boston
LOCATION
Boston Marriott Copley Place
DATE AND TIME
11/10/19 5:00pm - 11/10/19 7:00pm
The American Porphyria Foundation will be hosting a Patient Education and Support Meeting in Boston, MA on Sunday, November 10. RSVP TODAY!
I'll be there!
Maybe
I can't make it
Interested in a Patient Education and Support Meeting?
If you are interested in the possibility of having an APF hosted Patient Education and Support Meeting in your area, please contact the APF: 866-273-3635
Get Involved
Research
Visit our Website
Contact Information
Is your contact information up to date?
Medical Moments
Monday - November 4, 2019 @ 06:30:00
MEDICAL MOMENT
Below are the most prevalent symptoms of acute porphyrias (AIP, VP, HCP, ADP):
- abdominal pain and vomiting
- constipation
-
pain in the back, arms and legs
- muscle weakness (due to effects on nerves supplying the muscles)
- urinary retention
- palpitations (due to a rapid heart rate and often accompanied by increased blood pressure)
- confusion, hallucinations and seizures
- insomnia
Sometimes the level of salt (sodium and chloride) in the blood decreases markedly and contributes to some of these symptoms.
See the APF website: www.porphyriafoundation.org
Physician Information Kit Available
Tuesday - October 15, 2019 @ 06:30:00
Upon request, the APF will send a comprehensive physician education packet on the acute porphyrias to physicians.
It includes educational materials on the diagnosis and management of the acute porphyrias; information on biochemical and genetic testing; Panhematin treatment for the acute porphyrias; and a recent article from the Annals of Internal Medicine on diagnosis and management of the acute porphyrias.
Please call our office at (301) 347-7166 to order the kit.
In addition, the APF will also send information on PCT and EPP to physicians upon request.
Please call our office at (301) 347-7166 to order the packet.
These packets are delivered via USPS, for physicians only.
THE FDA APPROVED SCENESSE!
Friday - October 11, 2019 @ 06:30:00
THE FDA APPROVED SCENESSE!
Clinuve Pharmaceuticals Ltd has announced that Scenesse/Afamelanotide 16mg has been approved in the USA
by the Food and Drug Administration for the treatment of Erythropoietic Protoporphyria.
The APF is proud of our EPP community who fought for years for this approval. There are 7000 known rare
diseases, 5% of them have an FDA-approved treatment. FINALLY, EPP is one of them! Congratulations on this monumental win.
Porphyria Post
Thursday - October 3, 2019 @ 06:30:03
October 3, 2019
Porphyria Post
Rare Disease Week on Capitol Hill
Travel Stipend Application
Submissions are being accepted for travel stipends to attend the 2020 Rare Disease Week on Capitol Hill! We would love to have the Porphyria community represented this year in Washington, DC. This event brings together hundreds of patients, patient advocates, industry leaders and more. Please submit your scholarship application today, as the portal will close on December 2, 2019.
Please note: Travel stipends are available up to $800.00. Registration for patients and patient advocates is free, however, you must pay for your hotel and travel costs out of pocket. The APF/EveryLife Foundation is not responsible for making these arrangements. Stipend recipients are required to attend the Legislative Conference on February 26th and the Hill Day on February 27th but may attend the other events as well. Limit one travel stipend per family can be awarded.
We need your help! The APF has created an extensive list of physicians around the world who are treating with people with Porphyria. This list helps when people from around the world contact the APF needing assistance finding a knowledgeable physician. YOU can help patients everywhere by sharing with us the name and location of your physician. Comment on this post, PM Edrin Williams, APF Director of Patient Services or call the APF office at 301.347.7166 to give us their information. Thank you!
Medical Moment: Molecular Diagnosis (DNA)
Monday - September 16, 2019 @ 05:30:01
Medical Moment: Molecular Diagnosis (DNA)
An international collaboration of porphyria experts is building an evidence-based database of verified pathogenic and benign variants in order to facilitate accurate diagnosis for the porphyria community, beginning with the three most common acute porphyrias (AIP, HCP, VP). A SPECIAL ARTICLE was recently published in Genetics in Medicine, a publication of the American College of Medical genetics and Genomics. This collaboration is among the European Porphyria Network (EPNET) of experts and the NIH-supported Porphyrias Consortium.
ABSTRACT: With the advent of precision and genomic medicine, a critical issueis whether a disease gene variant is pathogenic or benign. Such is the case for the three autosomal dominant acute hepatic porphyrias (AHPs), including acute intermittent porphyria, hereditary coproporphyria, and variegate porphyria, each resulting from the halfnormal enzymatic activities of hydroxymethylbilane synthase, coproporphyrinogen oxidase, and protoporphyrinogen oxidase, respectively. To date, there is no public database that documents the likely pathogenicity of variants causing the porphyrias, and more specifically, the AHPs with biochemically and clinically verified information. Therefore, an international collaborative with the European Porphyria Network and the National Institutes of Health/National Center for Advancing Translational Sciences/ National Institute of Diabetes and Digestive and Kidney Diseases (NIH/NCATS/NIDDK)-sponsored Porphyrias Consortium of porphyria diagnostic experts is establishing an online database that will collate biochemical and clinical evidence verifying the pathogenicity of the published and newly identified variants in the AHP-causing genes. The overall goal of the International Porphyria Molecular Diagnostic Collaborative is to determine the pathogenic and benign variants for all eight porphyrias. Here we describe the overall objectives and the initial efforts to validate pathogenic and benign variants in the respective heme biosynthetic genes causing the AHPs.
The APF asked our Facebook friends for their top questions they would ask a porphyria expert.
The following questions were submitted to Dr. Wang for his responses Q. Does EPP give us bad teeth? Also, do people with EPP get stomach pains or is that with the other porphyias? A. The porphyrin that accumulates in EPP patients is protoporphyrin IX, which does not cause discoloration to teeth or abdominal pain.
The type of porphyria that leads to discolored teeth is Congenital Erythropoietic Protoporphyria. The porphyrias that lead to episodic abdominal pain attacks are the acute hepatic porphyrias. Q. I have EPP and I have a severe reaction on my hands and lips. Do I seek urgent care? Also, what can you even do when you burn your lips? A. The acute reactions to sunlight in EPP can be very severe and, unfortunately, there are not many effective options to treat the symptoms. Nonsteroidal anti-inflammatory drugs (NSAIDS such as ibuprofen) and stronger pain medications like opiates often provide only partial relief. These symptoms generally resolve spontaneously within hours to days.
Q. Do children under 10 years of age really have AIP attacks? I have seen this on social media, and to my knowledge it starts in teenage years with hormones. A. Acute porphyria attacks occur when the body, and in particular the liver, has increased demand to make heme that the mutated enzyme cannot keep up with. Hormones are known to be important inducers of heme production by the liver. This is why more than 90% of the patients who have acute porphyria attacks are female, and also why the vast majority of the attacks occur during the age range when they are actively menstruating.
But there are other causes that can induce the liver to make more heme, so it is possible, though rare, for acute attacks to occur prior to the start of menses. I would recommend carefully looking for other causes of acute, episodic abdominal pain in younger kids in order to not miss other more common causes.
RESEARCH. IF NOT YOU, THEN WHO?
Thursday - September 12, 2019 @ 14:15:19
RESEARCH. IF NOT YOU, THEN WHO?
If you have ever asked a question about your type of porphyria only to have a physician respond with I dont know,you are a candidate to participate in research. The American Porphyria Foundation is actively recruiting research volunteers for all types of porphyria. We are calling on our patient members to boost the number of volunteers that participate in these studies. The government grants that fund rare disease research, through the National Institutes of Health, support a limited number of disease groups like porphyria. A critical area for assessment to fund a rare disease group is the number of participants. If there is not proof of steady growth in the number of research volunteers, we risk losing critical funding that supports our Porphyria Centers across the US. To secure ongoing funding, we urge you to participate. It is easier than you may think! We understand that it is an important and personal decision, but if not you then who??? If you are interested in volunteering for any of these studies, please contact Edrin Williams, Director of Patient Services, at edrinw@ porphyriafoundation.org for additional information. Some available opportunities: Longitudinal Study (All Porphyrias) Harvoni Study (PCT) Panhematin Prevention Study (AIP, VP and HCP)
1.866.APF.3635
Porphyria Post
Tuesday - September 10, 2019 @ 05:30:03
DR BRUCE WANG SELECTED AS WINNER OF BEST PRESENTATIONAT ICPP
Dr. Bruce Wang, Porphyria Expert at UCSF and American Porphyria Foundation Protect the Future physician, has won Best Presentation at ICPP for his excellent work titled Single Cell Transcriptomic Analysis of Hepatocytes in a Mouse Model of Porphyria Cutanea Tardain collaboration with B. Patkar, D. Burhan, and J. Phillips. 118 abstracts from 27 countries were submitted for presentations and posters. Congratulations Dr. Wang!
INTERNATIONAL CONGRESS OF PORPHYRINS AND PORPHYRIAS (ICPP)
With over 500 attendees from 59 countries, the ICPP meeting held in Milan, Italy, September 8-11, was a global success. The congress began with a Patient Day, where patient advocacy groups collaborated on our work and participated in lectures. The scientific program was three days of presentations from scientists, researchers and clinicians. The APF will share detailed information and key learnings from the Congress over the next several weeks.
GLOBAL PORPHYRIA ADVOCACY COALITION FORMALIZED
GPAC has been formalized. The APF is proud to be a founding member of the Global Porphyria Advocacy Coalition, an umbrella organization for porphyria patient advocacy groups worldwide. Work will focus on access to treatment, support, research, awareness and education of all the porphyrias. We are stronger together!
TWO TRUTHS AND A LIE EPP
Friday - September 6, 2019 @ 05:30:13
TWO TRUTHS AND A LIE
Christina Verkest I am a 27-year-old from the suburbs of Detroit. I was diagnosed with EPP when I was about 5 or 6 at Childrens Hospital in Downtown Detroit. Since I have dealt with my EPP for the majority of my life, I am very comfortable with it, and even when I dont feel as fashionablein the summer sun, I know how to own it and am happy to educate others about why I might be wearing long pants and sleeves in the middle of a sweltering July day. So I try to this bring this own it, even the not-so-great stuffattitude into my high school Spanish classroom, as almost all of them can relate in some way. I always spend time at the start of the school year ensuring that my classroom environment is one that allows students to feel comfortable, accepted and supported. So one of the ice breakersI like doing is the infamous 2 truths and 1 lie,where I share with my students two truths about my life and one lie. They have to guess which of the three is my lie. I like to get really obscure with these because it makes an impression on the kids, but it also allows me to be real with them about who I really am. One of my truths is I am a modern-day vampire.That usually catches their attention! After many of them guess that I am lying about being a vampire, I tell them that, in fact, I am! I take the time to explain that I have a rare genetic disorder which causes me to experience photosensitivity or pain when I am in the sun. I even paint them the picture of my space suit,which refers to the long pants, sleeves, hat and sometimes gloves that I have to wear when I go to amusement parks or the beach. While sharing this information at such an early point in the school year may require me to get somewhat vulnerable with the students, my hope is that I can educate them about the disease but also spread some awareness about the unique characteristics every one of us has.
ICPP Conference and Patient Day
Thursday - September 5, 2019 @ 06:00:02
ICPP Conference and Patient Day
LOCATION Milan, Italy
DATE Patient Day: September 8, 2019 Conference:September 8-11, 2019
Will we see you there?
Welcome to the AFP!
Wednesday - September 4, 2019 @ 10:51:22
Welcome to the AFP!
Thursday, August 29, 2019
Please share in a warm welcome to Iany Schneider, our new Administrative Assistant at the APF. Iany comes with non-profit experience in the healthcare realm along with a positive, contagious attitude! You can reach Iany at iany@porphyriafoundation.org.
She waiting for your call. 1.866.APF.3635
Medical Moment: Pain Awareness Month
Wednesday - September 4, 2019 @ 10:48:32
Medical Moment: Pain Awareness Month
The U.S. Pain Foundation has designated September as Pain Awareness Month. The APF will be sharing pain stories, featured articles, and information related to pain in the porphyrias, whether it is the neurovisceral pain of the acute porphyrias or the burning pain of the cutaneous porphyrias. In fact, two APF members will be featured on the US Pain Foundation website as their videos were selected among the top 30 nationwide (thank you Candace Johnson and Claire Richmond!)
Take a moment to read the attached article: Patient Perspectives on Acute Intermittent Porphyria with Frequent Attacks: A Disease with Intermittent and Chronic Manifestations.According to this study, Pain was usually the first symptom cited by patients with AIP when asked to define porphyria or describe their experience of the disease. It was the most common and distressing symptom experienced by patients, both during attacks and chronically.
Also of interest, The Department of Health and Human Services organized a task force including multiple agencies to develop guidelines for patients and physicians. The APF made public comment and many patients shared their pain stories with this committee.
The U.S. Pain Foundation has designated September as Pain Awareness Month.
Tuesday - September 3, 2019 @ 23:35:46
Medical Moment: Pain Awareness Month
The U.S. Pain Foundation has designated September as Pain Awareness Month. The APF will be sharing pain stories, featured articles, and information related to pain in the porphyrias, whether it is the neurovisceral pain of the acute porphyrias or the burning pain of the cutaneous porphyrias. In fact, two APF members will be featured on the US Pain Foundation website as their videos were selected among the top 30 nationwide (thank you Candace Johnson and Claire Richmond!)
Take a moment to read the attached article: Patient Perspectives on Acute Intermittent Porphyria with Fr Medical Moment: Pain Awareness Month equent Attacks: A Disease with Intermittent and Chronic Manifestations.According to this study, Pain was usually the first symptom cited by patients with AIP when asked to define porphyria or describe their experience of the disease. It was the most common and distressing symptom experienced by patients, both during attacks and chronically.
Also of interest, The Department of Health and Human Services organized a task force including multiple agencies to develop guidelines for patients and physicians. The APF made public comment and many patients shared their pain stories with this committee. The Pain Management Best Practices Inter-Agency Task Force final report can be found here: https://www.hhs.gov/sites/default/files/pmtf-final-report-2019-05-23.pdf.
Update from CLINUVEL on US FDA REVIEW PROCESS SCENESSE
Tuesday - September 3, 2019 @ 23:33:18
Update from CLINUVEL on US FDA REVIEW PROCESS SCENESSE
Thursday, August 22, 2019
Ongoing interactions between the US Food and Drug Administration (FDA) and CLINUVELs scientific teams continue to take place as the final outcome date of October 6, 2019 approaches. This date has alreayd been extended from May 31st to October 6th. In the latter half of the scientific review, the FDA will attempt to address substantive review issues which have not yet been covered by its committee members. At this stage, the FDA will use its time to gain a broader insight of risks and benefits while reserving the right to identify new review issues that may trigger an extension of the Prescription Drug User Fee Act (PDUFA) goal date. There is no indication that the current PDUFA date cannot be met; it is CLINUVELs expectation that October 6th will remain the current PDUFA goal date for an outcome to be provided.
The Food and Drug Administration (FDA) hosted the American Porphyria Foundation on Monday, August 26th for a Rare Disease Listening Session. Patient Listening Sessions are a way for the FDA to engage with patients or their advocates and they are one avenue for a patient community to share their experience with a disease or condition by talking directly with FDA staff. The objective of the meeting with the APF was to identify porphyria as eight distinct diseases that fall in two main categories (acute and cutaneous), to share patient perspectives on living with different types of porphyria and to convey our desperate need for treatment for all our members. Dr. Amy Dickey gave an overview of the porphyrias to set the stage for the patient talks. We were proud to have seven patients share their experiences living with Porphyria. Thank you to Jason Barrett, Jennifer Beck, Amy Chapman, Gudron Debes, Amy Dickey, Desiree Lyon, and Morgan McKillop for representing the APF community. Each person did an excellent job sharing their message. The FDA responded with specific questions and clarifications related to various types of porphyria and with the feedback that we had met our objective for this meeting.
Patient Education Programs
Thursday - August 22, 2019 @ 14:27:36
Patient Education Programs
The American Porphyria Foundation is dedicated to the health and well-being of individuals affected by Porphyria. One way that the APF does this is by Patient Education Programs
Maintain, update, and expand comprehensive website, brochures, pamphlets, books and educational materials for each type of porphyria, genetics and research.
Develop new educational programs and services for purposes of research, treatment, diagnosis.
Distribute materials on porphyria treatment
Engage and assist in Health Insurance assistance and billing problems
Update relevant new information
Update on RESEARCH- ALL THE TIME
Participate in convention exhibits
Develop & deliver patient education programs for physician relationships
Educate the FDA drug approval process
Educate on congressionional process
Promote research process and participation
Arrange conference calls with experts
Provide expert consultation with patients and physicians
Members,
We cant do this alone. If you need any type of help or services please call the APF, we are happy to assist you! 1.866.APF.3635
ENEWS **IMPORTANT**
Friday - August 16, 2019 @ 06:30:00
ENEWS- **IMPORTANT**
Update from CLINUVEL on US FDA REVIEW PROCESS SCENESSE Ongoing interactions between the US Food and Drug Administration (FDA) and CLINUVELs scientific teams continue to take place as the final outcome date of October 6, 2019 approaches. This date has alreayd been extended from May 31st to October 6th. In the latter half of the scientific review, the FDA will attempt to address substantive review issues which have not yet been covered by its committee members. At this stage, the FDA will use its time to gain a broader insight of risks and benefits while reserving the right to identify new review issues that may trigger an extension of the Prescription Drug User Fee Act (PDUFA) goal date. There is no indication that the current PDUFA date cannot be met; it is CLINUVELs expectation that October 6th will remain the current PDUFA goal date for an outcome to be provided. Read Clinuvel's August Newsletter at https://www.clinuvel.com/â?¦/20â?¦/08/CLINUVEL-Communique-VI.pdf
Medical Moments
Acute Porphyria Medical Moment: Do you know the benefits of Prophylactic Heme Treatment? Read the abstract from an article written by Porphyria Expert, Dr. Herbert Bonkovsky. Attachment to the full article included below. Acute intermittent porphyria (AIP), an autosomal dominant inborn error of metabolism, is the most common and severe form of the acute porphyrias. Attacks of severe abdominal pain, often with hypertension, tachycardia, are cardinal features of AIP, often requiring hospital admissions. Frequent recurrent attacks of AIP, defined as > 3 attacks in one year, during which at least one attack requires intravenous heme therapy, are associated with significant morbidity, lost productivity, and health care burden. We report two patients with such frequent attacks of AIP, who have been managed with prophylactic heme therapy on a weekly basis. We describe results particularly in relation to symptom control, biochemical findings, health care costs, quality of life, and utilization of resources. During 11-month duration of weekly prophylactic heme infusions, we observed a 100% decrease in acute attacks and inpatient admissions in one subject and a 75% decrease in the other. During this time, we also observed a significant decrease in the number of emergency room visits. The decrease in number of acute attacks requiring hospital admission was associated with significantly decreased health care costs and improved quality of life. Reduction of both emergency room visits and hospital admissions decreased the utilization of health care services. Outpatient weekly infusions were also noted to be associated with better reimbursements and reduced overall costs of health care for the subjects. Both our subjects also endorsed better symptom control, quality of life and better understanding of disease. Thus, prophylactic heme therapy, through a multi-disciplinary approach, decreases the incidence of acute attacks, decreases health care costs and leads to better patient satisfaction and quality of life. Link: https://www.researchgate.net/â?¦/Benefits-of-prophylactic-hemâ?¦
PCT Medical Moment: Hepatitis C According to experts - Hepatitis C is common in PCT. In some areas where this viral infection is quite prevalent, especially in southern Europe and some parts of the U.S., as many as 80% of PCT patients are infected with this virus. How this particular virus contributes to developing PCT is not known. Other hepatitis viruses are seldom implicated.If you have Hepatitis C, have PCT and you are interested in participating in research, please contact Edrin Williams, Director of Patient Services for information about the Harvoni Study!
CEP Medical Moment: Do you have our CEP Informational Page? This page can be shared with your caregivers and healthcare providers as an educational tool. Please click on the link below to download! If you would like me to send you a hard copy via mail, please email me at edrinw@porphyriafoundation.org - Edrin Williams, Director of Patient Services. Link: Please add attached document (CEP One Pager)
Find Your Shadow 2019
Last week the APF sent the McKillop Family to the Happiest Place on Earththrough our Shadow Jumpers program! This deserving family of five was able to take advantage of all the parks had to offer VIP style! With thoughtful planning, Morgan (EPP) was able to stay protected from the sun while having a once in a lifetime experience. Morgan especially loved the thrill rides and took total advantage of the disability fast pass, which reduced her waiting times in the sun and reflective pavement. The family also had a special VIP tour guide, Meagan, who ensured that Morgan was taken care of and always positioned out of the sun. Though the weather was mostly overcast, during the sunnier times the family was able to shadow jump through stores, they only had to break out the sun umbrella ONCE! The family suggests taking mid-day breaks in order to avoid the worst of the sun and regroup. The APF was overjoyed to hear all about the McKillops vacation and thrilled to have been a part of making it happen! We are already looking forward to Find Your Shadow 2020!
101 Acute Hepatic Porphyrias 3 of 3
Wednesday - August 14, 2019 @ 06:30:07
Treatment During Attacks
Pain can be the primary and only complaint of a patient in a porphyric crisis and sometimes for some time beyond. Appropriate pain measures should be applied and it would be unwise and not good medicine to overrule a patient by withholding medication.
Hospitalization is often necessary for acute attacks. Medicine for pain, nausea and vomiting along with close observation are required. Initial treatment of AIP attacks consists of stopping harmful drugs the patient may be taking and providing a high intake of carbohydrate (300 grams or more per day). Carbohydrate can be given either in the form of an oral carbohydrate or by intravenous infusion. Intravenous infusion is better in moderate or severe attacks or for patients who are unable to ingest enough carbohydrate orally. Pain, anxiety and emotional symptoms should be treated with safe drugs. Attacks with muscle weakness occasionally require respiratory support, but this is unusual unless an attack is brought on by prolonged administration of harmful drugs. After recovering from an attack, a patient should continue to eat regularly, because there is good evidence that skipping meals or fasting is harmful.
Heme therapy is a treatment for AIP that is given intravenously. It is useful during severe attacks. The best results are achieved if heme therapy is started early in the attack. It is also administered to prevent attacks that are readily predictable. Heme therapy is commercially available through Recordati Rare Diseases as Panhematin. Panhematin is less likely to produce phlebitis if it is mixed with human albumin before it is given. Directions for preparing Panhematin in this manner can be obtained from Porphyria specialists (and are available from the APF as part of the ER Kit). Panhematin therapy is seldom indicated unless the diagnosis of acute Porphyria is proven by a marked increase in urine PBG. How heme therapy should be used to prevent attacks is not well established.
Heme arginate, which is marketed in some other countries, is another preparation of heme therapy for intravenous administration, but it is not available in the United States.
AIP is particularly dangerous if the diagnosis has not been made and if harmful drugs are continued. The prognosis is usually good if the disease is recognized and if treatment and preventive measures are begun before severe nerve damage has occurred. Although symptoms usually resolve after an attack, some patients develop chronic pain. Nerve damage and associated muscle weakness can improve over a period of months or longer after a severe attack. Long term expert physical therapy and other rehabilitation therapies are essential. In addition, mental symptoms may occur during attacks but are usually not chronic.
AIP patients prone to attacks should eat a normal or high carbohydrate diet and should not greatly restrict their intakes of carbohydrate and calories, even for short periods of time. If weight loss is desired, it is advisable to consult a physician who may then request that a dietitian estimate an individual's normal caloric intake (this varies greatly from one person to another). Then it may be appropriate to prescribe a diet that is approximately 10% below the normal level of calories for the patient. This should result in gradual weight loss and usually will not cause an attack of Porphyria.
Pregnancy is tolerated much better than was formerly believed. Offspring have a 50% chance of inheriting the gene for AIP, but the great majority of those who inherit the gene remain "latent" for all or most of their lifetimes.
Patients with frequent intermittent symptoms should have access to physicians who are familiar with AIP and with the patient's specific medical problems. Psychiatric support is sometimes helpful as well, because emotional problems may continue even with appropriate medical treatment. If new symptoms arise, diseases unrelated to Porphyria should be suspected. Like anyone else, AIP patients may develop other illnesses.
Precautions
Most individuals who inherit AIP seldom have symptoms if certain precautions are taken. The following general recommendations are made for individuals with low PBG-D blood test results:
Avoid harmful drugs, smoking, alcohol, hormones, and fasting. Make sure you and your physician review your medications for safety before you take them. Both of you should feel free to consult one of the experts in Porphyria and the APF drug database for this purpose since information about drugs and Porphyria is difficult to find.
Inform all of your physicians that you have AIP. You should know whether your urine is usually positive for PBG. Many physicians are not experts on Porphyria, thus, it is helpful for patients with AIP to carry information about themselves. A Medic Alert card and bracelet or necklace are also recommended for those who are susceptible to AIP attacks. In case unexpected emergencies occur, they could prevent harmful drugs from being administered.
Review requirements for surgery with your physician before the procedure. Surgery should be done without barbiturate anesthesia. The anesthetic gases are probably safe in AIP. Since major surgery interferes with nutrition, at least 300 grams of glucose should be infused intravenously during surgery and on a daily basis for a period of time thereafter.
Check urine periodically for PBG, particularly in children at the time of puberty. If the urine does become positive for PBG, most individuals still remain asymptomatic. However, since AIP symptoms are almost always associated with high PBG output, it is useful to have such information for future reference.
Contact Porphyria specialists or medical centers that have expertise in Porphyria in case unforeseen questions arise concerning drugs, treatments and other matters.
Be your own best advocate by educating yourself about Porphyria. Challenge your health care providers to also become very knowledgeable. It is essential to transfer from any health care providers who refuse to acknowledge the importance of this disease.
Seek medical attention immediately when you feel ill with anything other than ordinary illnesses.
Please note: the rapid PBG test kit is no longer available
Clinical Features of AIP
Signs and symptoms of AIP usually occur intermittently and include abdominal pain, constipation, muscle weakness, pains in the arms and legs, insomnia, emotional difficulties, rapid pulse, and high blood pressure. Muscle weakness can be severe during a prolonged attack due to the effects of Porphyria on nerves that control muscle. In fact, all symptoms of AIP appear to be due to effects on peripheral nerves, the nerves in the abdomen or the central nervous system. Precisely how Porphyria produces pain and other symptoms related to the nervous system is not yet well understood. PBG is produced in excessive amounts by the liver in AIP, but it has not been proven that excess PBG can damage nerve tissue.
Because Porphyria is rare and its symptoms often suggest other diseases, the correct diagnosis is often delayed. Attacks due to harmful drugs commonly occur before AIP is recognized. Symptoms of Porphyria can develop in some people but not in others who inherit AIP. If they occur, symptoms are characteristically intermittent. Some reasons for this variability are as follows:
Certain drugs are harmful to Porphyria patients. This is likely to occur if drugs are given for other illnesses or at a time when surgery is needed.
Hormones are clearly important in activating Porphyria. Quite a number of facts suggest this relationship. Porphyria patients metabolize (or break down) hormones differently than do normal subjects, and these hormone break-down products can cause the liver to make more PBG and porphyrins. In children who inherit AIP (detected by the PBG-D test), the condition is latent before puberty. After puberty, AIP may become active in some of these children.
Attacks are more common in women who inherit AIP than in men. Attacks in women often occur during the second half of each menstrual cycle, suggesting that female hormones are more significant than male hormones in bringing AIP from the latent to the active state. Attacks can be produced by oral birth control pills that contain synthetic types of female hormones.
Some women with AIP, on the other hand, have fewer attacks of Porphyria after certain types of birth control pills are started. This is thought to be due to a reduction in the rate of formation of their own female sex hormones, as a result of their taking the synthetic hormones in birth control pills. Thus, such synthetic hormones may, at times, be helpful. Some women have benefited from using a low-dose estrogen patch.
Nutrition is another important factor that can alter the course of an AIP patient. Fasting, a marked restriction of calories or a low carbohydrate diet can cause an attack. A high intake of carbohydrate is beneficial to shorten an attack when it occurs. Most AIP patients do well between attacks on a normal diet. A person with AIP who wishes to lose weight should do so gradually. For more detailed information, you may order the Diet and Nutrition brochure from the APF, or visit the Diet and Nutrition section of this website.
Environmental factors such as chemicals of various types, may play a part in predisposing a patient to increased symptoms of AIP. Some studies suggest that the chemicals that are found in cigarettes, insecticides and weed killers have this potential.
Alcoholic beverages should be avoided.
Other factors may precipitate or exacerbate an AIP attack such as stress of illnesses unrelated to Porphyria, extreme emotional stress or physical fatigue.
Drugs and AIP
Certain drugs are extremely important in exacerbating AIP. Patients with AIP are often treated with harmful drugs such as tranquilizers and sedatives before the disease has been diagnosed. This may lead to a severe attack. Porphyria will improve greatly after these drugs are stopped. Severe and fatal attacks are almost always related to the use of harmful drugs. This is one very important reason for testing blood relatives of known Porphyria patients to determine if they have the genetic defect.
Harmful drugs
Recommendations about drugs in AIP are based on specific test results and experience in patients with Porphyria in whom attacks have been caused by drugs. Since most new, commonly used drugs have not been tested, they should be avoided if at all possible. If a question arises, a physician or medical center specializing in Porphyria should be contacted.
The most important harmful drugs are barbiturates and sulfonamides (sulfa drugs). Barbiturates are commonly used as tranquilizers, sleeping pills and general anesthetics. Sulfa drugs are antibiotics commonly used to treat kidney and bladder infections.
Did you know that the APF will send a complimentary physician education packet to any of your treating doctors? Primary Care, Internist, Family Medicine, Hematologist, Oncologist, Gastroenterologist, etc..
If you want all your physicians invested in your care, let us help you educate them about porphyria. These complimentary packets are tailored to be specific to your type of Porphyria. As an example, the acute porphyria packets include information about the intensity of acute porphyria pain. The EPP packet includes information on the visible spectrum of light triggering phototoxicity. Email Edrin Williams, Director of Patient Services to request your packet(s) to be sent today! Please include their name and physical location.
Please note that the symptoms and treatments for AIP are applicable to Variegate Porphyria (VP), Hereditary Coproporphyria (HCP), and ALAD Porphyria (ADP). Unlike AIP and ADP patients, however, VP and HCP patients often develop photosensitivity.
Acute Intermittent Porphyria (AIP) is a rare disease that is usually inherited from one parent. It is almost always latent (i.e. does not produce illness) in childhood and is usually latent in adults throughout life. When the disorder is active, it can cause intermittent attacks of abdominal pain as well as a variety of symptoms, which range from mild to life-threatening.
After the correct diagnosis of AIP is made, simple precautions can be taken to prevent attacks. It is important to check all relatives of Porphyria patients for the genetic defect, so they can take the same precautions and avoid becoming ill from Porphyria. Even relatives who have never had symptoms should be tested, as described below.
Diagnosis
Laboratory tests are the keystone of diagnosis in AIP. Because signs and symptoms are nonspecific, test samples should be handled with care and sent to a laboratory of excellence for testing and subsequent evaluation. A laboratory overseen by a Porphyria specialist would be the best choice.
Blood tests
The enzyme porphobilinogen deaminase (PBG-D) is abnormally low in the liver and in most other tissues of a patient with Acute Intermittent Porphyria. This is true whether or not the patient has clinical symptoms or biochemical activity of the disease. Activity of this enzyme can be measured fairly easily in red blood cells. This is a useful test, particularly for evaluation of relatives of previously diagnosed patients with known low levels of PBG-D. This enzyme was formerly known as uroporphyrinogen-1-synthase (URO-S), a term still used by some laboratories.
However, there are some practical difficulties with the red cell assay:
In about 10% of patients and families, there is no decrease in activity of this enzyme, because the enzyme in red blood cells is slightly different from that in liver and most other tissues. Such patients and families have a defective enzyme expressed only in liver and other tissues, not in their red blood cells.
Even when the enzymatic defect is expressed in red blood cells, about 10% of patients' values fall into a zone that overlaps with that of the normal population. This is referred to as the "indeterminate zone".
Care must be exercised in the handling and storage of blood samples for testing. Although the PBG-D enzyme is relatively stable, activity will fall if samples are not refrigerated promptly and kept frozen until the time of assay.
Urine tests
Urine may appear purple during an attack or after standing in the light
The amount of porphobilinogen (PBG) in urine is increased during attacks of AIP. PBG is a porphyrin precursor, not a porphyrin. The Watson-Schwartz test or the Hoesch test is used in many hospital laboratories as a qualitative test of the urine to determine whether there is an increased amount of PBG. The result is reported as "positive" or "negative", but the actual amount of PBG is not measured. Measuring the amount of PBG in a 24-hour specimen or even a spot sample of urine is a much better test. A very high urine PBG, when determined by a reliable method such as the Mauzerall-Granick method, is diagnostic for the presence of an acute Porphyria.
There are three acute porphyrias that can cause increases in PBG, namely AIP, Hereditary Coproporphyria (HCP) and Variegate Porphyria (VP). Acute attacks can occur in all of these conditions. Skin photosensitivity can occur in HCP and VP, but not AIP.
There are several considerations to keep in mind regarding PBG measurements:
Urine PBG usually remains high between attacks of AIP, especially if the attacks are frequent. Thus, although a high PBG does indicate that the patient has an acute Porphyria, it does not prove that the patient is having an attack at the time.
Urine PBG may gradually become normal if there have been no attacks of AIP for a long time.
Urine PBG can become normal within 1-2 days after heme therapy.
In patients with HCP or VP, urine PBG can become normal more quickly after attacks than in patients with AIP. Urine porphyrins usually stay increased in these patients even when the PBG falls to normal.
Urine PBG is normal in most individuals who have inherited AIP but have never had symptoms. Therefore, it is not reliable for testing relatives.
Urine porphyrins are usually increased in acute porphyrias, but measuring them is less useful than measuring PBG. Increases in urine porphyrins are common in other medical conditions. Therefore, finding an increase in urine porphyrins may not mean that the patient has Porphyria.
The cost of care for rare medical conditions can place a significant financial burden on families. The following resources may help you find the financial support you need.
Does the National Institutes of Health (NIH) provide financial assistance?The NIH is the nations medical research agency. The NIH is not authorized to provide direct financial assistance but can provide medical care to participants who are enrolled in a clinical research study.
In the following sections, you will find nonprofit, government, and community resources that can help with financial assistance. We also encourage you to search the GARD website using the name of your condition. On the GARD disease pages you can find resources in the Organizationsand Living Withsections that may be able to provide financial assistance.
Are there any organizations that can provide financial assistance for medical costs related to rare diseases? Needy Meds is a nonprofit organization that lists programs that help people who cannot afford medications and healthcare costs. NeedyMeds has information about government programs, low-cost or free medical and dental clinics, and prescription assistance. NeedyMeds also has disease-specific financial aid programs. You can contact them directly at 800-503-6897.
The Pharmaceutical Research and Manufacturers of America (PhRMA) created a tool that allows patients to search for financial assistance resources offered through various biopharmaceutical industry programs to help cover the cost of medications.
The Patient Advocate Foundation (PAF) provides case management assistance for the uninsured or underinsured with life-threatening or debilitating illnesses. Services include help with the following: access to care; co-pay assistance; social security disability applications; and insurance appeals. PAF also has a National Financial Resource Directory that allows patients to find resources within a given state. You can contact PAF directly at 800-532-5274.
Family Voices aims to achieve family-centered care for all children and youth with special health care needs and/or disabilities. Family Voices has a map to help you find the Family-to-Family Health Information Center in your state. You can contact them directly at 888-835-5669.
The National Organization for Rare Disorders (NORD) is a patient advocacy organization for individuals with rare diseases and the organizations that serve them. NORD provides Patient Assistance Programs that provide financial assistance for medications, insurance premiums and co-pays, diagnostic testing, and travel for clinical trials or consultation with disease specialists. In addition, NORD provides links to other financial assistance resources. You can contact them directly at 800-999-6673.
The Assistance Fund provides various services to help patients with chronic or serious illnesses cover the cost of FDA-approved medications. You can contact them directly at 855-845-3663.
Good Days provides help to patients with specific life-altering conditions. Assistance includes help with the cost of medications and travel. You can contact them directly at 877-968-7233.
Are there resources for those with cancer who need financial assistance? The National Cancer Institute, part of the National Institutes of Health (NIH), has general information for Managing Costs and Medical Information. NCI also has a list of more than 100 organizations that provide emotional, practical, and financial support services for people with cancer.
CancerCare Co-Payment Assistance Foundation helps people cover the cost of co-payments for chemotherapy and targeted treatment drugs. You can contact them directly at 866-552-6729.
The Dental Lifeline Network uses a national network of volunteer dentists to provide dental care to those with a financial need who are 65 or older, disabled, or medically fragile. You can contact them directly at 303-534-5360.
NeedyMeds has a feature that allows individuals to find low-cost/free/sliding scale dental clinics in their geographical area. You can contact them directly at 800-503-6897.
How can I learn more about fundraising? Online fundraising, also known as crowdsourcing, is another method to consider when faced with financial hardship because of a rare disease. Some patients find that friends, family, and community members are willing to contribute financially if they are aware of a difficult situation.
Global Genes has created toolkits that provide individuals with information about a variety of topics related to living with and/or advocating for a rare disease. They have one toolkit called 5 Essential Tips and Tools for Effective Fundraising. Where can I learn more about dealing with insurance companies? Global Genes has a booklet titled Navigating Health Insurance.
The Patient Advocate Foundation has a pamphlet titled Insurance Denials & Appeals that may also be helpful. How can I learn more about government programs that provide financial assistance?There are state and federal programs to help people who cannot afford medical care or prescription drugs. USA.govprovides links to federal resources that help with various expenses, including medical care. You can contact them directly at 844-872-4681. You can find federally funded free or low-cost medical and dental care from the US Department of Health and Human Services Health Resources and Services Administration (HRSA).HRSAs Maternal & Child Health Bureau helps to increase access to quality health care and services for Americas mothers, children, and families.
Making Home Affordable is a government website designed to help individuals who are behind in their mortgage payments or unsure how to make the next payment. You can contact them directly by calling 888-995-HOPE.
Where can I learn more about applying for disability benefits? The Social Security Administration (SSA) defines disability on the basis of a persons inability to work and not on the specific medical condition. The SSA has information about How to Qualify for Social Security Disability Benefits.
The SSA also has a Compassionate Allowances Initiative that speeds up the processing of disability claims for applicants with certain medical conditions that cause severe disability.
How can I learn about community resources? You may find it helpful to speak with your state or county health department or social workers at your local hospital to request more information on available resources. Religious service organizations and community volunteer agencies such as the Salvation Army, Lutheran Social Services, Jewish Social Services, Catholic Charities, the Kiwanis Club, the Rotary Club, and the Lions Club may offer help. Many of these organizations are listed in your local phone directory. Some temples, mosques, churches, and synagogues may provide financial help or services to their members or those in need in their community. 211 Information is a confidential service provided by United Way Worldwide. The service provides people with local information about human services for everyday needs such as food, housing, and health. This service also provides resources in times of crisis.
How can my story make a difference? Many people with a rare or genetic disease struggle with medical costs. Families USAhas created a form to encourage people to share their experiences of dealing with the healthcare system. They are looking for stories of success as well as stories that illustrate the need for further improvements in the healthcare system. Families USA is a national nonprofit organization that works to promote high-quality, affordable health care for all Americans.
Are there resources for those who live outside the United States? If you live outside the United States and need help with financial assistance for a rare medical condition, GARD information specialists have a list of Patient Advocacy Groups located in other countries.
EURORDIS is a patient-driven alliance representing over 700 rare disease patient organizations in Europe. EURORDIS may have disease-specific resources that can help with the cost of living with a rare disease.
Orphanet is a European reference portal for information about rare diseases and orphan drugs. Orphanet allows you to find patient advocacy organizations that support specific rare diseases in different countries. Click the Patient Organisationstab at the top of the specific disease page.
Trying to find an underlying diagnosis for many conditions can be a very long and frustrating experience. With more rare conditions, a diagnosis can often take many years. Although this can be incredibly difficult, the following information may help you navigate through the process of trying to obtain a diagnosis.
Where can I find out how to cope with an undiagnosed condition?To learn more about how to deal with genetic or rare conditions that have no definitive diagnosis, see:
Are there research programs available for people without a diagnosis? Yes. If an individuals health care providers and specialists have not been able to make a definite diagnosis so far, participating in a research study or clinical trial may be another option. See below for a description of some of the National Institutes of Health (NIH) research programs that are going on now:
The Undiagnosed Diseases Network (UDN) is a research study funded by the National Institutes of Health Common Fund. The UDN is made up of clinical and research centers across the United States working to improve diagnosis and care of patients with undiagnosed diseases. Physicians and patients with additional questions may call 1-844-746-4836 (1-844-Ring-UDN).
ClinicalTrials.gov is database that provides current information on clinical research studies. You can search ClinicalTrials.gov for research studies looking at general categories of diseases (e.g. neurological diseases or eye diseases) or specific symptoms. Some studies accept individuals without a diagnosis with the research goal of making a diagnosis.
One study that is enrolling individuals who do not have a diagnosis is entitled "Pediatric patients with Metabolic and Other Genetic Diseases". This study is evaluating individuals with known or suspected genetic diseases, including metabolic diseases. Despite the name, people of all ages may be eligible for this study.
To find out more about clinical trials that take place at the NIH, you can call the NIH Clinical Center to talk to a specialist.
Patient Recruitment and Public Liaison Office NIH Clinical Center National Institutes of Health Bethesda, Maryland 20892-2655 Toll-free: (800) 411-1222 Fax: (301) 480-9793 E-mail: prpl@mail.cc.nih.gov
How can I learn more about clinical trials? If you or someone you know is interested in enrolling in a clinical trial, you can find helpful general information on clinical trials at ClinicalTrials.gov.
Resources on many charitable or special-fare flights to research and treatment sites and low-cost hospitality accommodations for outpatients and family members, as well as ambulance services, are listed in the GARD Help with Travel Costs guide.
Are there any advocacy groups for people with an undiagnosed condition? Yes. See below for additional information and supportive resources for individuals with an undiagnosed condition and their families.
Syndromes Without A Name (SWAN) is a supportive organization for families of children who have undiagnosed, unnamed conditions, or who are still looking for a diagnosis.
Syndromes Without A Name (SWAN) United States Toll-free: 888-880-SWAN Telephone: 269-692-2090 E-mail: info@swanusa.org Web site: http://swanusa.org/
The National Organization for Rare Disorders (NORD) is a federation of more than 130 nonprofit voluntary health organizations serving people with rare disorders. The NORD Web site includes information on medication assistance programs and networking programs, a resource guide, and links to other online resources. You can get this information through NORD's Web site or by calling or writing the NORD offices.
National Organization for Rare Disorders 55 Kenosia Avenue PO Box 1968 Danbury, CT 06813-1968 Toll free: 800-999-6673 (voicemail only) Telephone: 203-744-0100 TDD: 203-797-9590 Fax: 203-798-2291 E-mail: orphan@rarediseases.org Web site: http://www.rarediseases.org/
NORD recognizes that patients and families coping with undiagnosed rare medical conditions may experience unique challenges in accessing appropriate medical care and social support. Undiagnosed patients include those who are not yet diagnosedbecause they have not been referred to the appropriate medical specialist as well as patients who have a condition not previously described and for which a diagnostic test is not yet available.
Who should I talk to if I have financial concerns? It can sometimes take many years of specialized appointments and testing for a condition to be diagnosed, and this affects many individuals and families financially.
The Patient Advocate Foundation is a non-profit organization that serves as a liaison between families and their insurer, employer or creditors to resolve insurance, job retention and/or debt crisis matters related to their medical conditions. You can contact the Patient Advocate Foundation for further information.
Are there organizations that can help with the cost of travel? Yes. Traveling to specialized centers for testing and diagnosis can be costly; the following organizations help organize free travel for patients within the US.
Where can I find out more about financial assistance? The National Organization for Rare Disorders (NORD) provides information on financial and medication assistance programs, health insurance, medicare/medicaid programs, and links to additional online resources. Most of these resources are available only to individuals in the United States.
Many individuals want to know about healthcare professionals or researchers who have knowledge of their conditions. When a condition is rare, it can be difficult to find someone who has seen many cases. Although there is no list of experts in rare diseases, the guidelines below include several ways to identify healthcare professionals who have experience with a particular condition. Potential resources include patient advocacy groups, researchers who have conducted or are conducting clinical trials, and authors of articles published in the medical literature.
We are providing these resources to assist you in your search; however, sometimes it will not be possible to find a healthcare professional who has extensive experience in a particular rare condition. At the bottom of this fact sheet, we have provided some suggestions to help you when you are still unable to locate an expert after researching these resources.
The GARD Information Center provides these resources for informational purposes only and not as an endorsement of services. You should use your own judgment when evaluating a healthcare professional. You can find helpful information on choosing quality health care from the Agency for Healthcare Research and Quality (AHRQ).
How can I find a healthcare professional with experience in a specific condition? Several resources may be able to assist in your search for a healthcare professional with experience in a particular condition:
Many disease advocacy organizations have medical advisory boards, physician locator services, or patient networks, all of which may help you find a healthcare professional who is familiar with a particular condition. You can search for a condition on this website to find related disease advocacy organizations. These would be located in the "Organizations" section. If you dont find a specific group, search the Genetic Alliance and the National Organization for Rare Disorders (NORD) websites.
Published resources provide another way to find a specialist for a particular condition. Experts are often called upon to contribute to online publications such as GeneReviews and NORD. Many of these resources list the authors name and institution and may provide an e-mail address or phone number.
You can also search the medical literature to find healthcare professionals or researchers who have published recent articles or case reports on a particular condition. You can find relevant articles through PubMed, a searchable database of biomedical journal articles. Although not all of the articles are available online for free, most articles listed in PubMed have a summary/abstract available. In addition, contact information for one of the authors may be listed. On the Results page, select "Abstract" under Display Settings to view information about the authors.
How can I find a specialty treatment center? Treatment centers often have healthcare professionals of various specialties who work together. The resources listed below may help you locate a treatment center for your condition.
Disease advocacy organizations often establish Centers of Excellence for their condition(s). Contact a support organization to determine if they know of a Center of Excellence.
The National Cancer Institute provides information on how to find a doctor or treatment facility if you have cancer (many cancers are rare).
The Muscular Dystrophy Association (MDA) provides medical services for its members through MDA clinics across the country. These clinics serve people with a range of neuromuscular disorders. Patients can receive medical care for genetic disorders at a discounted cost through the MDA clinics. To learn about accessing these services, contact the MDA toll-free at 1-800-572-1717.
How can I find a genetics clinic?Genetics clinics are a source of information for individuals and families with a genetic condition. To find a genetics clinic, we recommend that you contact your primary doctor for a referral. Learn more about genetic consultations.
The following online resources can also help you find a genetics professional in your community:
The National Society of Genetic Counselors provides a database of genetics counseling services, searchable by location, name, institution, type of practice, or specialty.
The American Society of Human Genetics (ASHG) is a professional organization of researchers and clinical geneticists. The ASHG maintains a database of its members, some of whom live outside of the United States. Visit the ASHG site if you are interested in obtaining a list of the geneticists in your country, though some may be researchers only and may not offer medical care.
How can I find a researcher who is studying my condition?Researchers who are studying a specific condition are another source for identifying an expert. You may want to look for researchers who are conducting a clinical trial, as they are often medical doctors. In addition to asking for more information on their research, you can also find out if they see patients who are not enrolled in a study. If a researcher does not see patients, you may consider asking if they know a colleague who could help you. Some researchers publish periodic updates on their discoveries, and it may be possible for you to receive the latest information about their research.
The Rare Diseases Clinical Research Network (RDCRN) is made up of 22 distinctive consortia that are working to improve the availability of rare disease information, treatment, clinical studies, and general awareness for both patients and the medical community. The RDCRN also aims to provide up-to-date information for patients and to assist in connecting patients with advocacy groups, expert doctors, and clinical research opportunities. Visit the RDCRN site to see if your condition is supported by one of the RDCRN consortia.
The Centers for Mendelian Genomics program is working to discover the causes of rare genetic disorders. This program is collaborating with healthcare professionals and researchers working with individuals who have Mendelian disorders. For more information about the Centers, please visit their website.
ClinicalTrials.gov was developed by the U.S. National Institutes of Health, through the National Library of Medicine, to provide patients, family members, and members of the public with current information on clinical research studies. Through this resource, you may be able to find researchers who are studying your condition. You can conduct a basic search using the condition name as your search term. After you click on a study, review its "eligibility" criteria to determine its appropriateness. Use the study's contact information to learn more. Check this site often for regular updates.
The Patient Recruitment and Public Liaison (PRPL) Office at the National Institutes of Health (NIH) provides information about participating in research being conducted at the NIH Clinical Center hospital. Call the PRPL toll-free at 1-800-411-1222, or send an e-mail to prpl@mail.cc.nih.gov, to contact a liaison who can help you determine if there are any open clinical trials that interest you. They may also be able to provide you with contact information for researchers involved in these trials.
The Genetic Testing Registry lists laboratories offering research genetic testing for many conditions. Much of this research is conducted by physicians and scientists with an interest in a particular disease. To see if there are researchers studying your condition, visit the link above and use the condition name as your search term. Click on the "Research" tab on the results page.
What if I cant find a disease specialist?You may want to consider contacting a doctor at a university health center in your area, since university health centers tend to have the latest technology and treatments. University health centers have doctors who are involved in clinical trials and who may work together with others to diagnose and treat patients.
MedlinePlus, a website designed by the National Library of Medicine to help you research your health questions, provides links to directories to help you find health professionals, services and facilities.
If you have further questions, call 1-888-205-2311 to speak with a GARD Information Specialist.
Last updated: 6/13/2019
Share this content:
Medical Moment: Patient/Physician Relationship
Tuesday - July 23, 2019 @ 06:00:08
Medical Moment: Patient/Physician Relationship
We all want a great relationship with our doctors, right? You, as the patient have a responsibility to establish a solid rapport with your physician and other members of the healthcare team. This can have a positive impact on the quality of care and better access to treatment. Yes, there could be a stigma associated with having Porphyria but dont let it stop you from receiving the proper treatment that you deserve. Communication is KEY! Below you will find some key elements and tips that will not only prepare you for your visit with your doctor, but also build a strong relationship.
Here are a few tips for your doctors visit:
1. Plan Be prepared! Prepare your questions and concerns beforehand. You want to be courteous of your physicians time with you.
2. Make a list Make a list of your questions, concerns and any other relating information.
3. Communication is key Make sure that you understand fully what the doctor is explaining/advising. If you do not understand, SPEAK UP!
4. Access to Care Toolkit Download our recently updated Conversation Tracker. This document is helpful to keep detailed records of your conversations and interactions with doctors, nurses, counselors, and any other healthcare staff.
What Is A Spoonie, And How Can The Concept Help You?
Have you heard someone with a chronic illness mention the word spoonie? This actually refers to an incredibly important metaphor that allows chronic patients to describe their illness and the challenges they face to another person. It can refer to any individual who suffer from a chronic illness. The term spoonieoriginated from The Spoon Theory, a blog post written by Christine Miserandino. Essentially, the Spoon Theory illustrates the challenges that someone living with a chronic illness faces every day. Each day, spoonies have a limited amount of energy and ability. This energy is represented by a handful of spoons. Activities of daily living take away spoons. This makes it harder for chronic patients to keep up with daily tasks and responsibilities.
In 2010, Christine Miserandino was a college student living with lupus. She and her best friend were sitting in a diner, chatting about life in general. When Christine pulled out medicine to take with her food, her friend asked what it was like to have lupus. Christine was shocked. She recalled her reaction:
I was shocked not only because she asked the random question, but also because I assumed she knew all there was to know about Lupus. She came to doctors with me, she saw me walk with a cane, and throw up in the bathroom. She had seen me cry in pain, what else was there to know?
As anyone living with a chronic illness can attest, taking medicine, crying in pain, or struggling through the simple tasks of life are hard enough. If the illness happens to be invisible,even good friends and family can have a hard time truly understanding what it feels like. Invisible illnesses are those that are not readily apparent and include things like fibromyalgia, lupus, chronic pain, depression or other mood disorders, and chronic fatigue (among others).
What does spoonie mean?
Christine realized that her best friend needed a concrete way to understand what her daily struggle was like. The Spoon Theory was born.
If you assume that the day starts with ten spoons, then a morning for someone working and living with a chronic illness might go like this.
One spoon
Wake up and get out of bed. This may be challenging because chronic pain makes an early start difficult due to poor sleep and the fact that many pain conditions are worse in the morning.
One spoon
Brush teeth and get ready for work. This should actually be more than one spoon, as there are numerous actions that make this incredibly challenging, including all of the bending and stretching involved (drying feet, holding one arm up for two minutes to brush teeth). Getting dressed is another challenge, especially if clothes have multiple buttons or difficult closures. Many people with chronic illness have to resort to easy-to-pull on clothes with zippers and no buttons.
One spoon
Make breakfast. Many medications for chronic illness must be taken with food, so breakfast is essential. Trouble is, with so much activity already, a spoonie may be feeling tired to the point of nausea already. If there are children in the house to feed before school or lunches to pack, this part of the day can consume many more than one spoon.
One spoon
Drive to work or doctors appointments. Those who dont have a chronic illness take for granted the act of driving, but a spoonie has to consider all steps involved, from actually getting into the car (Is the seat too high? Then lifting their bodyweight into the car. Too low? Easing into the drivers seat engages muscles that may already be exhausted) to sitting up straight and focusing on the road. While medications may ease symptoms, they can also cause drowsiness, making attention difficult.
One spoon
Walk into appointment or work. Those with invisible illnesses are often eligible for handicapped car tags but may choose to forgo those. Because there are no crutches, canes, walkers, or wheelchairs, a spoonie may be subjected to nasty looks from those who see them park. Rather than deal with that, spoonies may decide to just park in the regular spaces and face a long walk to their desk.
The above example is a conservative view of what a morning with a chronic illness might look like. In reality, many spoonies use up all ten of their daily spoons in the first few hours of their day. They can borrowspoons from the next day, pushing to get through, but they will pay for it in fatigue.
Put even more simply, spoons represent energy. And using those spoons has a real consequence for people with chronic conditions.
Who is a spoonie?
Healthy people have a theoretically unlimited supply of spoons every day and can take this for granted. Those living with a chronic illness know that their spoons are limited. They must plan their day accordingly, conserving what energy they have when they can. That way they can focus on what is most important for that day. It is important to understand that this dance with the spoons is an ongoing, daily occurrence. There is no break or holiday for those with chronic illness or pain. Spoonies cannot just do it.Whatever itis must be planned and prepared for, sometimes days or weeks in advance.
The good news about the Spoon Theory and being a spoonie is that it can help to not only explain to people exactly what life is like with chronic illness but also to empower the person with a chronic condition. It may be difficult to always have to budget energy. This model though can help a spoonie plan their day around the things that are most important.
Especially for those with a spouse and children, this model can help make clear what a spoonie can and cannot accomplish in any given day. This might also encourage family members to pitch in and take over tasks that might be necessary but not crucial for a spoonie to complete. For example, even the youngest family members can make their own breakfast, help load a dishwasher, and pack their lunches. If the bus stop for school is in sight, developing a ritual of kisses, goodbyes, and waves at the door instead of a walk all the way to the bus might be an option. Every spoon-saving little bit helps.
Why is the spoon theory so important?
Having a solid metaphor explain what life is like with chronic illness can help you share more with your loved ones. But it can also help you understand how chronic illness affects your life.
Again, in Christines words:
[Its] hard, the hardest thing I ever had to learn is to slow down, and not do everything. I fight this to this day. [I] hate feeling left out, having to choose to stay home, or to not get things done that I want to. [Spoon theory helps loved ones] feel that frustration. [It also helps loved ones] understand, that everything everyone else does comes so easy, but for me it is one hundred little jobs in one. I need to think about the weather, my temperature that day, and the whole days plans before I can attack any one given thing. When other people can simply do things, I have to attack it and make a plan like I am strategizing a war. It is in that lifestyle, the difference between being sick and healthy. It is the beautiful ability to not think and just do. I miss that freedom. I miss never having to count spoons.
Chronic pain patients, especially, get really good at hiding their pain and finding ways to cope. Sometimes sharing how it affects you can help others better help you. It can also combat the social isolation many chronic pain patients feel. It can help them connect with other patients who feel the same.
How to find other spoonies online
Naming the Spoon Theory can help. But, using social media with the term spoonie and the hashtags #spoonie and #spoonieproblems can be an even greater support. Finding other spoonies online can help those with invisible illnesses to connect to others just like them. In the past, there has been a stigma behind not looking sick, but as more peoplereveal their struggles, that stigma may be lifting slowly, with growing understanding and acceptance.
You can find other spoonies on any social media site, but especially Twitter or Tumblr. You can also find many people who use the spoon theory to describe their pain in our chronic pain support group on Facebook. Having someone to talk to about your illness and the challenges it creates in your life can be incredibly therapeutic. Spoonies united can also help create a real impact in the way we talk about people with chronic pain and other conditions.
You can also find other spoonies talking about their chronic illness on YouTube. Annie Elainey is a vlogger who talks about the Spoon Theory on her channel.
Other communities of spoonies exist around the self-care packages that Spoonie Essentials releases. Opening up your box each month with other Instagram users is a fun way to talk to other spoonies. Bonus! You get to find products that help your everyday life.
Meet Shadow Jumper Mitchell Felts
Wednesday - July 17, 2019 @ 11:06:08
Meet Shadow Jumper Mitchell Felts
Mitchell Felts, age 12
How old were you when you were diagnosed? I was 9 Years old.
Do you remember your first flare/reaction? No, I do not recall my first flare & reaction.
What did it feel like to you? It was tingling bad on my skin.
What things help you feel better? (cool water, ice, shade, bath, clothes) It helps me when I use cold rags and ice packs on my skin.
How long does it take before you start to feel better? Sometimes, I start to feel better after the first day, but sometimes it takes 2-3 days or longer to feel better.
What kind of clothing/trends do you wear when you go outside or in bad lighting? I can use long sleeves, hats and umbrellas.
What is your most favorite sport to play or watch? Do you play it inside or outside? During the day or at night? I enjoy playing baseball late in the day and it feels great to play at night.
What ways are you able to adapt to do certain activities outside? I must cover up, its hot outside so I must take breaks to cool off and other times I just must stay inside.
What fun things are you able to do inside your home while the sun is out? I love to play on my Play Station.
What has been the best vacation ever? The Ocean, I did have a few attacks along the way but it was worth it.
Have you met any other kids with EPP before? I was able to meet a few kids with EPP in Washington D.C.
Do you go to School/Homeschool? I go to School.
Do you have to go by car, bus or walk? I go in the car.
What things do you have to do to protect your skin? I use my sleeves and hats to keep protected from the light/sunlight.
Is it hard for you to tell your friends, teachers or family what EPP is? Yes, it is hard for me to talk to them about it. I dont think they could understand.
What things do you do if you feel sad or left out? I play my games inside the house.
What do you want to be/do when you grow up? I want to be an Army or Police Officer.
Parents- How do your children deal with EPP? He mentions that hes fine and hides the pain until he is unable to handle it anymore. Most days he stays inside and periodically goes out on the front porch to see if he can handle the sun, so he can play outside. For the most part hes embarrassed of this disease and tries to hide his pain and suffering.
Would you benefit from a new treatment? He has had no treatment! It would be nice for him to be able to leave the house without his emergency bag (sunblock, different size hats, umbrella, sleeves, plastic bags for ice, sunglasses and extra shirt). It would be nice to go to any event and not check for shade and the closest place to get ice. It would be nice to not worry about vitamin D, Calcium, Iron and liver enzymes. For him to go outside to play and not worry about a reaction to the sun and him breaking another bone would be wonderful.
Thank you Mitchell & Cassie for explaining how you feel about your EPP & how your able to cope when you have a bad reaction. To learn more about EPP read more about it @ www.Porphyriafoundation.org
Amazing patient advocacy.and needed media for this issue. Thank you Terri Witter!
Wednesday - July 17, 2019 @ 06:00:11
Amazing patient advocacy.and needed media for this issue. Thank you Terri Witter!
We know growing up with EPP has its challenges. You may have to cover up and look different outside or may be stuck indoors on those hot sunny days but whats important is to know that you arent alone out there. Many kids, just like you, are dealing with long days finding things to do to dodge the sun and ways to let loose at sun down. Here at Shadow Jumpers, weve developed a section for Kids to hear from people just like YOU! Check back here soon to read some amazing testimonial stories from kids in our EPP community. EPP may be hard, but its something we continue to tackle together.
I wear an oversized hoodie the hood covers the sides of my face. (Brady, age 12)
Cut thumbholes in your sleeves to keep your jacket or sweatshirt over your hands without sliding up. This works great for young kids! (anonymous)
Wear a big hat and walk on the shady side of the street! (Brenda, EPP)
I have a bag prepared with a long-sleeved shirt or light jacket, hat, gloves, and face cover (I use a bandanna, some use a buff) that I take with me on cloudy days so If it clears up Im covered and dont put myself in DANGER. (Rob, EPP)
Our son wears a zip-up lightweight jacket with a hood in the summer so that more he can unzip it to get more air and stay cooler.
Buy inexpensive sports gloves (for example, at Five and Below). We cut the tips off the fingers so that he can still manage things with his hands.
QUICK SHADE!:
Use a taller person as shade (Rob, EPP)
Walk next to buildings for shade (anonymous)
Tell teachers, coaches, other parents, etc. that THEY are your childs quickest protectionâ?¦just stand between them and the sun! (Kristen, caregiver)
Hold their hands to protect them from the sun especially when you dont have any gloves.
Always put your back to the sun.
SCHOOL:
Meet with teachers at the beginning of each year to explain EPP and offer guidelines for safety (caregiver)
Initiate a 504 plan for your child. (Ask the APF for a copy of a sample plan!)
Make sure your teachers know that your child needs a safe place for fire drills, recess, physical education class and field trips.
DRIVING:
Carry a blanket or towel in your car at all times and close it in the top of the window to provide shade. (Kristen, caregiver)
Have your car windows tinted with as dark as possible tinting that is allowed in your area.
VACATIONS:
We always bring along a canopy on vacation. We can set it up wherever our son is to provide him some safe shade. (EPP caregiver)
THINGS TO DO INSIDE:
So often during the summer I would go to the movies while all my friends were away during the day at camp. From the popcorn to the cool temperature to combat the hot summer days, I would spend hours at a time throughout the week at my local theater.The downside? So many movies meant so many tickets which meant so much money spent.Thats why, with this new thing called MOVIE PASS, going to the movies has been more affordable. For one flat monthly fee of $9.95, movie passer customers can see one movie a day each day for the month. To sign up and too see theaters in your area qualify go to https://www.moviepass.com/
THINGS TO AVOID:
Remember that water and snow reflect the sun!
Sun comes through windows, too.
UV INDEX APPS:
Hey Shadow Jumpers! Heres a cool tip I wanted to pass alongâ?¦ (Craig, EPP)
People with EPP often check the UV index every day. I know I regularly check the UV index for the day to know what I should wear or think about doing. So often people traditionally check the newspaper or kids rely on their parents. Today I wanted to share with you some great UV Apps!
Wolfram Sun Exposure Reference App Based on your skin type, what SPF youre wearing and the UV forecast for your location, this comprehensive app can predict exactly how long you can stay in the sun before burning. Organizing a trip to the shore? Check out a five-day UV forecast displayed on a map, and find out what hours each day you should minimize sun exposure. ($0.99; available for iOS)
Ultraviolet ~ UV Index Keep things simple with this cool tracker, which displays the current UV index in your area using a large, vibrantly colored circle. Blues and greens mean youre in the clear while reds and purples mean a dangerously high index. General sun safety advice will tip you off for when its time to put on a hat, apply sunscreen or avoid going outside altogether. Just the bare necessities for when youre bare at the beach. (Free; available for iOS)
EPAs SunWise UV Index The U.S. Environmental Protection Agency designed this easy-to-navigate application, which delivers location-based UV index information. Ideal for plan-ahead types, the most useful feature is the color-coded hourly forecast that makes it easy to spot when the UV index is highest. (Free; available for iOS and Android)
(PLEASE NOTE: EPP reactions occur from visible light. UV Index applications and apps will ONLY indicate ultra-violet light, not the visible light range that affects EPP)
APF Swag Store
Monday - July 15, 2019 @ 06:00:00
Check out the remaining Inventory of APF Swag! Get your's today! 100% of your purchase goes back to the APF.
Sizes & Colors left are in description! BOGO items!!
https://www.porphyriafoundation.org/apf-store/
Prices of shipping per country vary please email Amy@porphyriafoundation.org for pricing
Misconceptions about living with a rare disease the community weighs in
A few weeks ago, we asked the community what some misconceptions were about living with a rare condition. The Mighty collected your responses and published them in the article below on February 28 for #RareDiseaseDay.
On World Rare Disease Day, groups and communities representing over 7,000 rare diseases gather together to unify their messages and support one another. With so many conditions, and new ones continuously being discovered, its sometimes hard for others to grasp what it really means to live with a rare condition, or care for someone whos rare.
The Mighty teamed up with the National Organization for Rare Disorders (NORD), a non-profit organization supporting individuals and families facing rare conditions through education, patient advocacy and research, and asked our communities, Whats one misconception about living with a rare condition?Their answers show that indeed, there are many who think its a much easier path than it is. The struggle to receive access to treatment and care, as well as a knowledgeable medical team is very real.
One misconception about rare disease is that people think doctors and researchers are lining up to study you, do research and help you and that doctors will know what to do with you. Nope, nope, and nope. There are no adult outcomes, longevity, or lifespan knowledge for my childrens disorder. We are literally figuring it out as we go along.Lindsey C.
When people say, I hope she gets better or I hope she gets healed.I know its very well-meaning, but when you have a genetic disorder, every cell in your body contains your mutation, so thats just simply not possible. I just smile now; Ive stopped trying to explain.Kristy L.
Some people think that people living with rare diseases have no value, and that we arent worth curing.Julianna K.
One misconception is that if I dont make it to a doctors appointment, Im not really sick. Sometimes the symptoms of my rare disease prevent me from making it to the doctor.Heather S.
Treatments are not the equivalent to being cured. It is frankly just treatment. I am still fighting. I am still sick. I will need ongoing treatment forever to live as stable a life as possible.Mandy F.
There is a misconception that having a rare disease gets easier the longer you have it. In reality, you get more fatigued and behind with your life.Patricia C.
Some believe that those who share the same rare disease can be treated the same, but that isnt the case. What works for one doesnt work for everyone.Angie G.
People that dont have to live day-to-day with multiple, rare, chronic and degenerative diseases like I do dont understand that its a full-time job, even though I am unable to work. Its a constant battle juggling multiple doctors appointments and trying to keep my symptoms under control. So unless theyve never walked in my shoes, they have no place to judge me.Amelia C.
It seems to me that folks assume rare diseases are coveredby insurance like any other condition. They are surprised to discover that the medicines I need the treatments, even the doctors, are as rare as hensteeth and six times as expensive.Carolle C.
That we dont have good days where we can pretend to be like everyone else. Just because we are in pain doesnt mean we dont enjoy going out with friends, shopping, or being social.AnneMarie G.
That the treatment for a much more common disease could also work on a rare disease, even if they only have one symptom in common, but completely different causes.Katherine O.
People think that if Im smiling and seemingly happy, Im not in pain. They assume Im all better.When in reality, Im dying on the inside and just want to go back to bed.Ali K.
Some think that I should be better after all the doctors visits. Its not easy to treat something that even your physicians dont fully understand.Mindy A.
Its a misconception that a diagnosis comes quickly.Karen R.
With a rare disease, its nearly impossible to get others to understand youre in pain if you cant show them.Christy H.
We arent on some kind of a stay-at-home vacation just because we have a debilitating rare condition.Tessie A.
Our society thinks it takes a special kind of person to care for someone with a rare disease or disability. Nope. It just takes a kind of person who displays basic human qualities and has a lot of love and patience.Elizabeth B.
You can live a joy filled life with a chronic illness. It doesnt necessarily mean that you feel good, but that you choose to be joyful.Michelle P.
Ive had people think that changing my food and/or diet will completely curemy rare disease, which is not the case.Jenn S.
Theres an assumption that all doctors know and learn about rare diseases. This is certainly not true.Sandra R.
Its frustrating when people tell me, But you look great!My response? Thanks! You dont see me on the days I look like my medical record.Dawn D.
People need to understand that we oftentimes dont outwardly look physically ill. We could be your neighbor, your nurse, your teacher. You walk past lots of rare disease patients every day and have no idea who they are based on appearance alone.Kaitti M.
I think a major misconception of rare disease is that a diagnosis is given from birth or sought after. This is not always the case. Some rare disease patients are born in perfect health and live years before they have a triggerto their disease. Literally overnight, families will have to live with massive changes. They will now learn to live with severe disability and the medical maze of rare disease.Kim S.
Dont think that we dont love what we have. Having a rare disease comes with pluses and minuses, but I wouldnt trade my life if given the choice. So many incredible people have come into my life due to my rare disease. The tough times I endured have made me a better person.Kendra G.
A misconception of living with rare disease is that all experiences are the same. Instead, those experiences of people living with rare disease are as diverse as the people who live with even the same rare disease. There may be similarities to symptoms and treatments, but pain, medications, therapies, appointments, lifestyles will differ depending on who is living with the rare disease. We are still a kaleidoscope of our race, nationality, ethnicity, gender, sexuality, age, class and ability. Telling our complete and unique stories is imperative to patient-centered medicine.Rebekah P.
Welcome to Shadow Jumpers! This is a page for EPP kids and parents by EPP kids and parents. Shadow Jumpers was created to help give kids with Erythropoietic Protoporphyria (EPP) and their families a place to learn about this rare disease, read tips and tricks learned over time and to hear from fellow kids. Through spreading awareness, fellow EPP interviews, tips to protecting yourself outside and some insight for parents, we hope all families living with EPP will look at this condition as a challenge they can overcome. Be sure to check out all the Shadow Jumper resources! Wed love to hear from you. Reach out to us anytime on shadowjumpers@porphyriafoundation.org!!
A main goal of Shadow Jumpers is to help kids living with Erythropoietic Protoporphyria do things they have always wanted to do but have not been able because of the sun. There are only several hundred diagnosed cases of this ultra-rare genetic disorder in the United States so we need to learn from and support one another! Pain from the sun combined with the mental discomfort kids can feel from having to cover up may lead kids to avoid certain activities, relationships and paths in life.
Shadow Jumpers wants to show (and remind) kids that they can still follow their dreams! Whether thats trying to play a sport outdoors, going on a family vacation, camping or whatever comes to mind, Shadow Jumpers wants to help. Click around to learn about some awesome EPP kids, how we are trying to do our part and how you can too!
Here are ways to connect with Shadow Jumpers!
This critical OpEd was published today about access to Panhematin. Please share with your community and your members of Congress!
Wednesday - July 10, 2019 @ 13:18:51
This critical OpEd was published today about access to Panhematin. Please share with your community and your members of Congress! Email-kristen@porphyriafoundation.org https://morningconsult.com/opinions/rare-disease-sufferers-fighting-both-their-condition-federal-bureaucracy/
NIH UPDATED INFORMATION ON ALL PORPHYRIAS
Tuesday - July 2, 2019 @ 16:20:04
Here is an updated NIH article please read and share with anyone who has questions. The following information may help to address your question:
What is porphyria? How is porphyria diagnosed? Is genetic testing clinically available for porphyria? How can I find a genetics professional in my area?
What is porphyria?
Porphyrias are a group of blood conditions caused by a lack of an enzyme in the body that makes heme, an important molecule that carries oxygen throughout the body and is vital for all of the bodys organs. Major types include ALAD deficiency porphyria, acute intermittent porphyria, congenital erythropoietic porphyria, erythropoietic protoporphyria, hepatoerythropoietic porphyria, hereditary coproporphyria, porphyria cutanea tarda, and variegate porphyria. The most common type of porphyria is porphyria cutanea tarda. Some of the symptoms of porphyria include blistering, swelling, and itching when the skin is exposed to sun. Other symptoms may also include pain, numbness or tingling, vomiting, constipation, and intellectual disability. There is no known cure for porphyria, but the various types have different courses of treatment, and may include bone marrow transplant.[1]
Most porphyrias are inherited conditions with either an autosomal dominant or autosomal recessive pattern of inheritance. However, some forms of porphyria can be caused by environmental factors such as infections or exposures to certain prescription drugs. Porphyrias caused by environmental factors are called sporadic or acquired porphyria.[2][3] Last updated: 3/22/2017
How is porphyria diagnosed?
Many of the signs and symptoms of porphyria are similar to those of other more common diseases. Also, because porphyria is rare, many doctors have not seen cases of the disorder before, making it more difficult to diagnosis. Because porphyria's signs and symptoms usually aren't distinctive, laboratory tests are required to make a definitive diagnosis and to determine which type of porphyria is involved.[4] If your doctor suspects porphyria, he or she may recommend the following tests:[4][5]
Urine test. If you have a form of acute porphyria, a urine test may reveal elevated levels of two substances: porphobilinogen and delta-aminolevulinic acids, as well as other porphyrins. Blood test. If you have a form of cutaneous porphyria, a blood test may show an elevation in the level of porphyrins in the liquid part of your blood (plasma). Stool sample test. Analysis of a stool sample may reveal elevated levels of some porphyrins that may not be detected in urine samples. This test may help your doctor determine your specific type of porphyria. Genetic testing may also be used to confirm the diagnosis.[5] Last updated: 11/4/2016
Is genetic testing clinically available for porphyria?
Yes. Genetic Testing Registry (GTR) lists laboratories offering clinical genetic testing for several types of porphyria. Clinical genetic tests are ordered to help diagnose a person or family and to aid in decisions regarding medical care or reproductive issues. Talk to your health care provider or a genetic professional to learn more about your testing options. Last updated: 11/4/2016
How can I find a genetics professional in my area?
To find a medical professional who specializes in genetics, you can ask your doctor for a referral or you can search for one yourself. Online directories are provided by GeneTests, the American College of Medical Genetics, and the National Society of Genetic Counselors. If you need additional help, contact a GARD Information Specialist. You can also learn more about genetic consultations from Genetics Home Reference. Last updated: 8/23/2016
We hope this information is helpful. We strongly recommend you discuss this information with your doctor. If you still have questions, please contact us.
Over the years, the experts of the Porphyrias Consortium ( https://www.rarediseasesnetwork.org/cms/porphyrias/) have developed an extensive list of publications. These articles have served to educate both patients and healthcare professionals. This lengthy list of publications includes the implementation of research studies, clinical trials, diagnosis, management, current and emerging therapies, etc. of the porphyrias.
Our team of experts continue to work on your behalf to provide you with the most up-to-date information as research continues. Listed below are a few of the go-to publications.
We invite you to click on the link below to see the full list of publications.
Annals of Internal Medicine - Recommendations for the Diagnosis and Treatment of the Acute Porphyrias
Benefits of Prophylactic Heme Treatment
Diagnostic Delay in Erythropoietic Protoporphyria
Hepatitis C, porphyria cutanea tarda and liver iron: an update
***General article not involving the Porphyrias and check the drug safety list***
Medicine & Health
Avoid the Sun If You Take These Drugs
Ronilee Shye, PharmD, BCGP, BCACP Roni Shye, PharmD, BCGP, BCACP is a licensed pharmacist in Florida, Ohio, and Pennsylvania. Posted on March 26, 2019
If youre itching for sun and cant wait for summer, its important to know beforehand that some of your medications could cause an unexpected problem. You may not be aware of this, but some prescription drugs can make you more sensitive to sunlight and cause your skin to burn more easily, a reaction known as photosensitivity.
What symptoms happen with photosensitivity?
If your medication has a warning to avoid sunlight or mentions photosensitivity as a possible side effect, dont ignore it. Photosensitivity is an abnormally high sensitivity to ultraviolet (UV) rays from the sun. This usually means that you could be more sensitive to sunlight and get sunburns more easily. It might not be the same kind of sunburn youre used to either. You may end up with a worse sunburn than usualeven a little exposure could mean a severe burn.
You could also become more sensitive to other light sources, including indoor fluorescent lights. The reaction to UV or fluorescent lights can cause itchy spots or areas of redness and swelling on patches of exposed skin.
These common medications can make you more sensitive to the sun:
Antibiotics, particularly tetracyclines like doxycycline and fluoroquinolones like ciprofloxacin Tricyclic antidepressants like amitriptyline and nortriptyline Older antihistamines like promethazine Griseofulvin, an antifungal medication Quinine and other antimalarial medications Acne medications like Accutane (isotretinoin) and Retin-A (tretinoin) Some cancer drugs Sulfonylurea drugs for diabetes like glyburide, glipizide, and glimepiride Hydrochlorothiazide (HCTZ) and other thiazide diuretics Some heart medications for arrhythmia, including amiodarone
How can I minimize my exposure to the sun?
To cut down on your sun exposure, first (and this may be obvious) avoid direct exposure to the sun. Youll also want to stay away from tanning beds as they can be as bad or worse than direct sun exposure. When you do spend time outside, wear sunscreen! Protective clothing like long sleeves, pants, hats, and sunglasses can help.
And if you do get a sunburn, try cool compresses, and call your PCP.
Medical Moment: Hepatitis C
Thursday - June 27, 2019 @ 06:00:03
PCT
Medical Moment: Hepatitis C
Hepatitis C is common in PCT. In some areas where this viral infection is quite prevalent, especially in Southern Europe and some parts of the U.S., as many as 80% of PCT patients are infected with this virus. This virus contributes to developing PCT in part by decreasing hepcidin levels that tend to increase hepatic iron and in part by other increases in oxidative stress. Other hepatitis viruses are seldom implicated (Porphyrias Consortium).If you have Hepatitis C and PCT, contact Edrin at the APF today for information regarding a clinical trial offering FREE Harvoni.
APF 1.866.APF.3635
Medical Moment: EPP Testing
Tuesday - June 25, 2019 @ 13:00:42
EPP
Medical Moment: EPP Testing
This ultra-rare disease is diagnosed by testing the red blood cells for the presence of abnormally high levels of protoporphyrin. These tests need to be performed in a specialized laboratory for accurate results. The specific test to order to diagnose EPP is called Erythrocyte Protoporphyrinlevels. There are only a select number of laboratories in the United States that can perform the complex analysis to diagnose EPP.
The labs that perform this testing correctly are:
University of Texas Medical Branch at Galveston, Texas
Mayo Medical Laboratories
It is recommended that these laboratories are used because they offer metal-free and zinc protoporphyrin, which is essential for diagnosis and monitoring of EPP.
Medical Moment
Wednesday - June 19, 2019 @ 07:00:02
Medical Moment: Panhematin Access
Are you lacking access to Panhematin? Does your health system refuse to pay for Panhematin?Is your physician aware of the importance of early treatment? We need to know.
Panhematin is the only FDA approved treatment for acute porphyria attacks, and we want to make sure that EVERYONE with an acute porphyria (AIP, VP & HCP) has access. Unfortunately, FDA approval doesnt ensure access. Patients have fallen victim of the bureaucracy inside the Center for Medicare and Medicaid Services (CMS), whose policies effectively discourage treatment with Panhematin in the hospital setting that is recommended by physicians.We want to help healthcare providers secure access to Panhematin at their preferred health facility.
We know patients who are being denied this treatment from some hospitals and directed to secure another healthcare provider. If this has happened to you or someone you know or care for, please contact us at the APF office. We could help by assisting you contact your local and state representatives and health advocacy organizations. We understand the debilitating effects of acute porphyria and hope these resources will help you secure access to Panhematin when you need it most.
Porphyria friends in Argentina have a center for diagnosis and treatment headed by Dr. Alcira Batlle. For more information , please see: http://www.qb.fcen.uba.ar/cipyp/
Our member, Rafael de la Torre from Buenos Aires shares his experience with PCT . To contact him, email: rdelatorreu@hotmail.com
In 2001 I went to consult the dermatologist about some blisters that appeared in one of my fingers. After asking me about others symptoms, he asked me to have an analysis performed by Dr. Alcira Batlle at the hospital in Buenos Aires. The result was Porphiria Cutanea Tarda.
The treatment was S-Adenosyl-L-Methione, 800 mg/ day, during 20 days, and simultaneously I received 100 mg Chloroquine, twice a week, until the levels of urinary porphyrins reached the controls. And I also had one Phlebotony of 500 ml of blood. The concentration of urinary porphyrins and plasma porphyrins was measured at the beginning of the treatment and then every week, during therapy. When normal levels were reached, chloroquine intake was suspended. Clinical recovery was paralleled with biochemical recovery. I have regular controls performed every year, and since then I have been fine, without any relapse.
With the porphiria diagnosis I went back to the dermatologist, and he told me that I have to consult one hepatologist, because I could have some liver damage. I did have, I had hepatitis C. I followed the treatment for about nine months with a combination of pills and Interpheron injections. At the end of this treatment, thanks God, I was healed.
Nowadays, I have controls performed every year for this disease, too.
Nora began having difficulty in the sun during the summer when she was three. After being outside, she would cry inconsolably and in pain, sometimes for hours. The backs of her hands in particular seemed to bother her, and she would tell us they "itched." However, there were no outward manifestations-no rash, no swelling, no marks. We were stumped, as were the doctors we visited. At their recommendation, we tried various solutions, including Sarna lotion, Claritin, and weaning on and off various sunscreens-none of these worked. As a last resort, a dermatologist prescribed Benadryl, which worked only because it knocked her out.
Although there was we could see, we knew something real was happening. Throughout that summer and the next two summers, Nora would ask to leave pool parties, play dates, and other fun occasions because the pain was just too much to take. We resorted to staying inside.
Finally, one weekend in Nora's fifth summer, she developed a triangular, flat, purple lesion on her nose. It didn't look like a bruise or a scab, and we took her in to see the pediatrician on call, who was not our primary doctor. Thankfully, he had treated a patient with EPP twenty years earlier and recognized the disease. He directed us to the APF website, where we found an almost perfect match to her symptoms. We got a blood test as recommended on the site, and confirmed that she has EPP. Our regular pediatrician has educated herself about the disease, and even managed to speak with the wonderful Dr. Mathews-Roth on the phone. Nora now has her liver function and porphyrin levels tested annually.
We are so relieved to have a diagnosis, and we are now able to manage around her condition. Thanks to the APF, we have found Lumitene, which Nora starts taking in April to prepare her for the summer. It hasn't been a perfect solution, but, if she stays in the shade, she can participate in most outdoor activities for at least some period of time. Hats and clothing with sunblock provide less relief, as the heat of the sun seems to penetrate them and bother Nora. We also rely on COTZ sunblock.
We are fortunate to live in New England, so Nora is generally fine mid-fall through mid-spring. We visited Disney in Florida in February this year and, even with a build-up of Lumitene, COTZ, hats and sun-protective clothing, the sun was still too strong for her. We were shocked, as the temperature never exceeded 60 degrees. It was a startling reminder of her condition.
Nora attends a wonderful small, independent school that has accommodated her condition. When she started kindergarten, we sent a note to the staff with a link to the APF website. In another stroke of luck, one of the staff members was aware of porphyrias in general and that helped us in our education efforts. Thankfully, Nora doesn't need any accommodations for indoor activities, but the school does allow her to "sit out" physical education activities if she is getting too much sun which, at age seven, she is able to fairly accurately determine for herself.
Nora doesn't play summer sports or attend outdoor camps, but we've found some great indoor alternatives for camp, including a drama camp, cooking camp, and reading camp. She did play spring soccer this year, and was fine until the last week or so. She sat out the last few games in the shade and cheered on her team. We had prepared the coaches in advance for this eventuality and found them to be quite supportive.
In all, we think (and hope) that Nora feels like a "regular" kid! Maria Gulluni (Nora's mom)
A personal note from the APF: In one of life's funny coincidences, the staff member at Nora's school who was familiar with porphyria is a friend of mine. I've known David for years, and his wife Judy, a doctor, is a dear friend. Judy was a medical resident, and was in the hospital with me when someone first suggested I be tested for AIP. After I was diagnosed, Judy learned about all of the porphyrias, and David learned about them through her and through me. So if you have porphyria, talk to your friends and family about it! You never know how you might be helping another patient along the way. Mira Geffner
ENEWS
Thursday - June 13, 2019 @ 06:00:02
ENEWS
Member Stories
ð??·Share your story! We want to hear from youâ?¦
The American Porphyria Foundation wants to invite you to share your story with the porphyria community. With permission, all stories will be featured on the APF Website. We will also feature select stories in our quarterly newsletter.
Stories should be about 500 2000 words and full of your personality. Make sure to include information about your specific porphyria type, your diagnostic journey and how this disease has impacted your life.
We will be sending out a small token of our appreciation to the first 5 submissions.
We look forward to receiving your stories. The APF office hears regular feedback that the stories listed on our site have helped them feel less isolated and work toward diagnosis. Thank you for having a hand in helping others through your stories and words.
Send your stories to Edrin at edrinw@porphyriafoundation.org
Smile, Shop, and Support
Did you know that you can support to the APF by simply shopping on Amazon? The AmazonSmile Foundation donates a portion of eligible purchases to the charity/organization of your choice.
ð??·To start giving to the APF click here. Under the Amazon search bar you will see the word SUPPORTING in orange, change the organization to reflect the American Porphyria Foundation. Then, you are ready to start shopping and supporting! By simply buying your normal amazon purchases through AmazonSmile you are impacting those affected by porphyria.
Click here to learn more about AmazonSmile https://smile.amazon.com/gp/chpf/about/ref=smi_se_rspo_laas_aas
Patient Education and Support Meeting- NYC
ð??·LOCATION
Icahn School of Medicine at Mount Sinai
DATE AND TIME
07/10/19 5:30pm - 07/10/19 7:30pm
1428 Madison Ave. 1st Floor Division of Medical Genetics New York, NY Room: AB 1-31 Signs will be provided for direction.
Here is our story on my son, Briggs. I am beyond grateful for the APF. Without the APF we may not be where we are today with a diagnosis.
At three years old, Briggs began having problems with sun exposure. His symptoms started in February of that year. His hands would itch, burn and sting. He would scream that bugs or monsters with pointed noses were trying to come out of his skin and that the ants were in his hands and feet. He did his best to try to communicate his pain. We went to our pediatrician as well as an allergist/immunologist. He had many sets of bloodwork and allergy testing. We were left without answers. He had no swelling, rash, or heat on the skin. We went to the beach in July. After one day of sun exposure he had a full-blown episode. Until this point, we didnt realize that the episodes could be worse than what we had seen up to that point. We had multiple nights where he was up until 4am screaming. After the day of sun, we allowed him no more, but his symptoms remained. We used fans, cool rags, water, ice packs, Benadryl, Tylenol, you name it and there was no relief.
Following this trip, we amped up the search for answers. We had moved to another town, so we started with a new pediatrician. From here we saw two different neurologists and another allergist/immunologist. Briggs had countless tests, and nothing came back with any clear answers as to what was causing his symptoms. Briggs was never symptomatic when we were in front of a doctor. I recorded episodes and would play them for each doctor. Over the fall/winter months, the episodes decreased and finally went away. We thought we were home free, and that it was just something odd that he went through and it was gone.
He turned 4 in November, and around March the symptoms returned. He was in the sun one day over spring break and he had another full-blown attack. My dad and I began to dig tirelessly on the internet over the next couple of days. In our previous searches, the symptoms from conditions that we came across were not a perfect fit. Most conditions we came across would present with a rash, swelling, heat, etc. and Briggs never had that. Finally, my dad came across a Dateline special titled Out of the Shadows.After seeing this, we were combing the internet for EPP. I went to the American Porphyria Foundation website and read the Member Storiesposted on EPP. For the first time, I felt like I was reading our own story. I contacted APF the next morning and they were such a fabulous resource. They provided a list of specialists, instructions for testing, and encouragement as we walked down this road. I read and printed a stack full of literature and took it to our pediatrician. She connected with a specialist out of state and he was tested. We received a call from the doctor confirming that Briggs tested positive for Erythropoietic Protoporphyria. I knew in my heart this is what it was, but having the confirmation was difficult.
At this point were only a week and a half post diagnosis. Weve purchased sun-protective clothing and are making the needed adjustments to ensure that he is episode free. Our goal is to be as creative as possible with how our family lives life to give him as normal of a life as possible. My daughter contracted bacterial meningitis at 7 days old, and from our journey with her, I learned that as parents and caregivers we must be advocates for those we love. With Briggs, we were aggressive in our search for a diagnosis. Some people passed us off as overreacting or were simply okay with not having an answer. If you are pre-diagnosis, I encourage you to not stop.
Medical Moment
Tuesday - June 4, 2019 @ 13:52:15
Medical Moment:
When discussing your specific type of Porphyria with your healthcare provider, how often do you give them the acronym of your type of porphyria? AIP, VP, HCP, EPP, PCT, CEP etc.
It is important that you verbally state the entire name of your disease with your provider, rather than giving them the acronym. This could reduce the amount of errors caused in medical records daily. The relationship with your physician is built on communication! Set the tone, educate them and be assertive. There is nothing more important than making sure that your care is top priority.
Medical Moments
Monday - June 3, 2019 @ 12:28:40
Acute Groups ------
Medical Moment:
In an individual with an acute porphyria, an acute attack can be brought on by certain drugs, hormones in women, environmental factors including chemicals of various types, nutrition including fasting and low carbohydrate diets, alcoholic beverages, medical and physical stress, and physical fatigue.
What factors precipitate an acute porphyria attack for you? Comment below.
------
Cutaneous Groups:
Medical Moment:
Sun sensitivity is the main symptom in CEP, EPP, XLP and PCT. VP and HCP, which are acute porphyrias, can also have blistering sun sensitivity. The degree of sensitivity to sunlight varies considerably. Patients with sun sensitivity have high levels of porphyrins in the blood plasma which, depending on the type of porphyria, have originated from the liver or the bone marrow. Ultraviolet light interacts with porphyrins in such a way as to damage skin tissue. In general, patients with CEP, EPP, XLP and PCT should protect themselves to prevent sun exposure and patients with VP or HCP should also protect themselves if this is one of their symptoms.
What do you do to protect yourself from the sunlight? Comment below.
New & Updated Brochures Free at porphyriafoundation.org- APF Store-Materials Order yours today
It was early April 1997, and I was truly enjoying my job as the guidance department chairperson of a suburban Philadelphia high school and my caseload of 10th thru 12th grade students. But the stresses of the workload were wreaking havoc on my health. I was always in pain and uncomfortable. The doctors were always prescribing stronger doses of pain medication or injection therapies. My hospitalizations were more frequent and for longer periods of time. My internist at that time insisted that I take a medical sabbatical for a year to give my body time to recover and to allow me the time for advanced testing. It was a hard decision to make, but I knew I was not physically or mentally able to give the 200% effort I gave to my job. So I decided to do what the doctor ordered.
Then on Memorial Day of that year, Larry and I were in a serious automobile accident which left me with severe abdominal pain and a wackier-than-usual neurological state. After a summer from Hell and the persistence of my doctor, the testing was done and his gut feelingthat I had porphyria was verified. After over thirty years of pain, frequent hospitalizations, misdiagnoses, and multiple surgeries, I was diagnosed with Acute Intermittent Porphyria in September 1997. Finding a specialist to handle my treatment plan proved to be easier than the doctor, Larry, or I could ever have imagined.
I had been with a neurologist, for over a year after I had passed out in a gynecologists office before a routine exam. He was treating me for migraine headaches and the neurological symptoms that I had developed following a hysterectomy. During a visit, Larry happened to mention the recent AIP diagnosis and asked if he knew of any physicians in the Philadelphia area who worked with porphyria patients. Imagine our surprised reaction when he informed us that he was currently treating two other patients with AIP!! I became his third.
He had been treating the two patients with a regimen of Panhematin and D10 with much success. I was admitted to the hospital in December of 1997 for a pic line insertion and Panhematin infusions. Dr. Bell closely monitored the process to see how well I tolerated the infusions and the D10. If all went well, I would be able to receive my treatments at home. And so l entered the next phase of my life.
I became a patient of Home Infusion upon my release from the hospital. My doctor oversees my treatment plan. The Home Infusion pharmacist assigned to my case and my primary Home Infusion nurse, Janice, manages my monthly treatments. Im on my third pharmacist, Jerry. All three have been outstanding! Janice has been with me through it all! Initially, I was receiving infusions of Panhematin once a day for five days out of every 28 days. Sort of mimicking a menstrual cycle even though Id had a hysterectomy. Over the years, we have experimented with the dosage amounts and number of infusions and days. The meds and supplies are delivered to my front door the Friday before my treatment is to begin. The Home Infusion nurse comes to my home to mix and administer the Panhematin and to assess my condition. They all have come to feel like family. I also receive D10 via a CADD pump for the entire treatment week. Fluid bag and pump are housed in a backpack designed specifically for that purpose. I have a chest wall infusaport, so Im home (not in the hospital) and Im mobile.
I returned to work on a part-time basis for the 1998-99 & 1999-2000 school years. However, the stress of the job requirements (I never got the concept part-time), a couple of setbacks, and the death of my Father in 2000 convinced me that I could no longer do the work I so enjoyed. I was able to take a disability retirement from the school district. Since then, Ive been slowly trying to get my life back on track. I dont want to jinx myself, but I have not had to be hospitalized for an attack in a long time! The Panhematin treatment seems to work for me. I dont believe I would be here today it werent for this drug.
European EPP Camp For Teenagers (Ages 7-17)
Tuesday - May 14, 2019 @ 13:22:57
European EPP Camp For Teenagers (Ages 7-17)
Welcome to the 1st summer camp for EPP teenagers. We gather together and live some beautiful adventures in the south of France. This camp is located in a very safe neighborhood. The camp is open for every EPP teenager and we welcome you all. Some Countries that have already participated in this event are from Switzerland, Denmark, France and now is open to the United States.
The porphyrias are inherited genetic conditions, which means that people with a porphyria have changes to certain genes that affect their bodys ability to regulate itself. When genes are copied, either to make new cells or to make a child, sometimes the body makes an imperfect copy. There can be little changes in the genes, called mutations, which can occur randomly. Sometimes these changes do not make any difference in how well the gene works, but other times they can keep the gene from working properly (referred to as mutations) and are disease causing.
In the porphyrias, these mutations are in the genes involved in a certain chemical pathway, called the heme biosynthetic pathway. Heme is a compound that the body needs to make hemoglobin and there are several steps to make this compound in the body. Each type of porphyria is caused by a defect in a specific enzyme in the heme biosynthetic pathway. Without these enzymes working properly, the body is not able to finish making heme and it causes a buildup of other compounds, called porphyrins. It is the buildup of different types of porphyrins that causes the types of porphyria.
Porphyria cutanea tarda (PCT) is a form of porphyria that primarily affects the skin. People affected by this condition generally experience "photosensitivity," which causes painful, blistering lesions to develop on sun-exposed areas of the skin (i.e. the hands and face). Skin in these areas may also be particularly fragile with blistering and/or peeling after minor trauma. In some cases, increased hair growth as well as darkening and thickening of the affected skin may occur. Liver abnormalities may develop in some people with the condition and PCT, in general, is associated with an increased risk of liver cirrhosis and liver cancer.[1][2] In most cases, PCT is a complex or multifactorial condition that is likely associated with the effects of multiple genes in combination with lifestyle and environmental factors. For example, factors such as excess iron, alcohol, estrogens, smoking, chronic hepatitis C, HIV and mutations in the HFE gene (which is associated with the disease hemochromatosis) can all contribute to the development of PCT.[3][1] Less commonly, PCT can run in families (called familial PCT). Familial PCT is caused by changes (mutations) in the UROD gene and is inheritedin an autosomal dominant manner.[4][1] Treatment may include regular phlebotomies (removing a prescribed amount of blood from a vein), certain medications, and/or removal of factors that may trigger the disease.[1][2]
This table lists symptoms that people with this disease may have. For most diseases, symptoms will vary from person to person. People with the same disease may not have all the symptoms listed. This information comes from a database called the Human Phenotype Ontology (HPO) . The HPO collects information on symptoms that have been described in medical resources. The HPO is updated regularly. Use the HPO ID to access more in-depth information about a symptom.
The resources below provide information about treatment options for this condition. If you have questions about which treatment is right for you, talk to your healthcare professional.
Management Guidelines
Orphanet Emergency Guidelines is an article which is expert-authored and peer-reviewed that is intended to guide health care professionals in emergency situations involving this condition.
The American Porphyria Foundation offers a document that includes information about porphyria, types, testing, and treatment with Panhematin. Click the "document" link above to view these guidelines.
If you need medical advice, you can look for doctors or other healthcare professionals who have experience with this disease. You may find these specialists through advocacy organizations, clinical trials, or articles published in medical journals. You may also want to contact a university or tertiary medical center in your area, because these centers tend to see more complex cases and have the latest technology and treatments.
If you cant find a specialist in your local area, try contacting national or international specialists. They may be able to refer you to someone they know through conferences or research efforts. Some specialists may be willing to consult with you or your local doctors over the phone or by email if you can't travel to them for care.
You can find more tips in our guide, How to Find a Disease Specialist. We also encourage you to explore the rest of this page to find resources that can help you find specialists.
Related diseases are conditions that have similar signs and symptoms. A health care provider may consider these conditions in the table below when making a diagnosis. Please note that the table may not include all the possible conditions related to this disease.
Conditions with similar signs and symptoms from Orphanet
Differential diagnosis is mainly variegate porphyria (see this term), diagnosis of which rests on evidence of the characteristic fluorometric peak in plasma.
Research helps us better understand diseases and can lead to advances in diagnosis and treatment. This section provides resources to help you learn about medical research and ways to get involved.
Clinical Research Resources
ClinicalTrials.gov lists trials that are related to Porphyria cutanea tarda. Click on the link to go to ClinicalTrials.gov to read descriptions of these studies.
Please note: Studies listed on the ClinicalTrials.gov website are listed for informational purposes only; being listed does not reflect an endorsement by GARD or the NIH. We strongly recommend that you talk with a trusted healthcare provider before choosing to participate in any clinical study.
Patient Registry
The Porphyrias Consortium is a team of doctors, nurses, research coordinators, and research labs throughout the U.S., working together to improve the lives of people with this condition through research. The Porphyrias Consortium has a registry for patients who wish to be contacted about clinical research opportunities.
Support and advocacy groups can help you connect with other patients and families, and they can provide valuable services. Many develop patient-centered information and are the driving force behind research for better treatments and possible cures. They can direct you to research, resources, and services. Many organizations also have experts who serve as medical advisors or provide lists of doctors/clinics. Visit the groups website or contact them to learn about the services they offer. Inclusion on this list is not an endorsement by GARD.
Right now an estimated 30 million Americans live with a rare disease. In the U.S., nearly 7,000 diseases are considered rare, and for most, no cure exists and few (if any) effective treatments are available, according to the National Organization of Rare Disorders (NORD).
From the exhaustion of a diagnosis journey to the feeling of being alone, those with rare diagnoses and their families face a lot of unique challenges that may be difficult for others to understand.
The Mighty teamed up with NORD to ask their Facebook community what they wish others understood about their experiences. Heres what they had to say:
1. Id be more than happy to educate you if you ask about [my condition] rather than question its existence.Katie DeMore
2. Most doctors have never heard or seen a patient with the disease.Nancy Reeder Martin
3. Smiling doesnt mean Im suddenly healed. It just means Im choosing to stay as positive through the pain as possible. Evan Mundine
4. Do not give advice to people with rare diseases. I know more about my disease than my own doctor does so please think before attempting to give advice. Brittney Peebles
7. Just because what youre experiencing doesnt fit into an easily diagnosable box doesnt mean you should be easily dismissed and overlooked.Megan Wirts
8. Fundraising is a big deal because government funding is scarce or nonexistent.Rebecca Brewster
9. I wish people wouldnt say , Ahhh, I hope you feel better soonlike its the flu! Lauri Morris
10. Nothing about rare diseases is simple not the diagnosis, not the daily care, not the long term. If you really want to know about my child, its not a one sentence answer.Elizabeth Grehl Breden
11. Physicians need to understand that we likely know more about our disease than they do. They need to actually listen to us! Be compassionate and understanding.Renee Walchak LEcuyer
12. It feels incredibly isolating to want to help your child and not be able to because even the doctors dont know whats wrong. To not have someone who understands because there is nobody like your child. To not be able to have a treatment plan because there is no diagnosis.Susie Stretton
13. People can just be a good friend and listen and be there for you; they dont have to try to relate by saying they know someone who has the same thing. Hence the word rare.Jill Ritchey
14. Sometimes you have a name that everyone knows (i.e. epilepsy), but a rare presentation of it (i.e. Lennox Gastaut syndrome) that means you dont qualify for studies and the meds dont work. Its a terribly isolating and frustrating place to be in medically, and the generic name doesnt give the correct picture to the public.Kelly Shaughnessy Morris
15. One in a millionmeans its possible. And those onesneed to count.Hailey Remigio
16. You may never really get a full diagnosis and/or prognosis.Jessica Taylor
18. It cant be fixed by a simple visit to the doctor. There isnt a drug or something that can be given to cure it. Its always there and without research it always will be.Sylvia Marsden
19. To be able to talk to and meet fellow patients and have a conversation about our disorder without having to try to explain what it is is extremely valuable.Neil Smith
20. Some of your coworkers, neighbors and friends who appear to be living normalproductive lives are also living with rare disorders. I bet most people know (often without realizing it) at least one person who lives with a rare disorder.Lisa McClellan Lucius
21. We enjoy the confused look on [your] faces when we tell [you] the name of our illness/es! Neuromye-what?!Helen Lear
22. Just because a doctor/pediatrician/ER staff hasnt heard of a disorder doesnt mean it doesnt exist.Brittany Lazechko Alley
23. Our caregivers go through so many sacrifices caring for us! Its a thankless job and I know I dont always say thank you,but I would not be where I am without them! Mary Lou Briggs
24. You will do and learn things you never thought possible. Hopemeans so much more since sometimes that is all you have.Nicole Vallier
Misconceptions about living with a rare disease the community weighs in
A few weeks ago, we asked the community what some misconceptions were about living with a rare condition. The Mighty collected your responses and published them in the article below on February 28 for #RareDiseaseDay.
On World Rare Disease Day, groups and communities representing over 7,000 rare diseases gather together to unify their messages and support one another. With so many conditions, and new ones continuously being discovered, its sometimes hard for others to grasp what it really means to live with a rare condition, or care for someone whos rare.
The Mighty teamed up with the National Organization for Rare Disorders (NORD), a non-profit organization supporting individuals and families facing rare conditions through education, patient advocacy and research, and asked our communities, Whats one misconception about living with a rare condition?Their answers show that indeed, there are many who think its a much easier path than it is. The struggle to receive access to treatment and care, as well as a knowledgeable medical team is very real.
One misconception about rare disease is that people think doctors and researchers are lining up to study you, do research and help you and that doctors will know what to do with you. Nope, nope, and nope. There are no adult outcomes, longevity, or lifespan knowledge for my childrens disorder. We are literally figuring it out as we go along.Lindsey C.
When people say, I hope she gets better or I hope she gets healed.I know its very well-meaning, but when you have a genetic disorder, every cell in your body contains your mutation, so thats just simply not possible. I just smile now; Ive stopped trying to explain.Kristy L.
Some people think that people living with rare diseases have no value, and that we arent worth curing.Julianna K.
One misconception is that if I dont make it to a doctors appointment, Im not really sick. Sometimes the symptoms of my rare disease prevent me from making it to the doctor.Heather S.
Treatments are not the equivalent to being cured. It is frankly just treatment. I am still fighting. I am still sick. I will need ongoing treatment forever to live as stable a life as possible.Mandy F.
There is a misconception that having a rare disease gets easier the longer you have it. In reality, you get more fatigued and behind with your life.Patricia C.
Some believe that those who share the same rare disease can be treated the same, but that isnt the case. What works for one doesnt work for everyone.Angie G.
People that dont have to live day-to-day with multiple, rare, chronic and degenerative diseases like I do dont understand that its a full-time job, even though I am unable to work. Its a constant battle juggling multiple doctors appointments and trying to keep my symptoms under control. So unless theyve never walked in my shoes, they have no place to judge me.Amelia C.
It seems to me that folks assume rare diseases are coveredby insurance like any other condition. They are surprised to discover that the medicines I need the treatments, even the doctors, are as rare as hensteeth and six times as expensive.Carolle C.
That we dont have good days where we can pretend to be like everyone else. Just because we are in pain doesnt mean we dont enjoy going out with friends, shopping, or being social.AnneMarie G.
That the treatment for a much more common disease could also work on a rare disease, even if they only have one symptom in common, but completely different causes.Katherine O.
People think that if Im smiling and seemingly happy, Im not in pain. They assume Im all better.When in reality, Im dying on the inside and just want to go back to bed.Ali K.
Some think that I should be better after all the doctors visits. Its not easy to treat something that even your physicians dont fully understand.Mindy A.
Its a misconception that a diagnosis comes quickly.Karen R.
With a rare disease, its nearly impossible to get others to understand youre in pain if you cant show them.Christy H.
We arent on some kind of a stay-at-home vacation just because we have a debilitating rare condition.Tessie A.
Our society thinks it takes a special kind of person to care for someone with a rare disease or disability. Nope. It just takes a kind of person who displays basic human qualities and has a lot of love and patience.Elizabeth B.
You can live a joy filled life with a chronic illness. It doesnt necessarily mean that you feel good, but that you choose to be joyful.Michelle P.
Ive had people think that changing my food and/or diet will completely curemy rare disease, which is not the case.Jenn S.
Theres an assumption that all doctors know and learn about rare diseases. This is certainly not true.Sandra R.
Its frustrating when people tell me, But you look great!My response? Thanks! You dont see me on the days I look like my medical record.Dawn D.
People need to understand that we oftentimes dont outwardly look physically ill. We could be your neighbor, your nurse, your teacher. You walk past lots of rare disease patients every day and have no idea who they are based on appearance alone.Kaitti M.
I think a major misconception of rare disease is that a diagnosis is given from birth or sought after. This is not always the case. Some rare disease patients are born in perfect health and live years before they have a triggerto their disease. Literally overnight, families will have to live with massive changes. They will now learn to live with severe disability and the medical maze of rare disease.Kim S.
Dont think that we dont love what we have. Having a rare disease comes with pluses and minuses, but I wouldnt trade my life if given the choice. So many incredible people have come into my life due to my rare disease. The tough times I endured have made me a better person.Kendra G.
A misconception of living with rare disease is that all experiences are the same. Instead, those experiences of people living with rare disease are as diverse as the people who live with even the same rare disease. There may be similarities to symptoms and treatments, but pain, medications, therapies, appointments, lifestyles will differ depending on who is living with the rare disease. We are still a kaleidoscope of our race, nationality, ethnicity, gender, sexuality, age, class and ability. Telling our complete and unique stories is imperative to patient-centered medicine.Rebekah P.
Right now an estimated 30 million Americans live with a rare disease. In the U.S., nearly 7,000 diseases are considered rare, and for most, no cure exists and few (if any) effective treatments are available, according to the National Organization of Rare Disorders (NORD).
101-About Porphyria from NIH
Friday - April 12, 2019 @ 12:00:06
About Porphyria
The porphyrias are a group of different diseases, each caused by a specific abnormality in the heme production process.
What is porphyria?
The porphyrias are a group of different diseases, each caused by a specific abnormality in the heme production process. Heme is a chemical compound that contains iron and gives blood its red color. The essential functions of heme depend on its ability to bind oxygen. Heme is incorporated into hemoglobin, a protein that enables red blood cells to carry oxygen from the lungs to all parts of the body. Heme also plays a role in the liver where it assists in breaking down chemicals (including some drugs and hormones) so that they are easily removed from the body.
Heme is produced in the bone marrow and liver through a complex process controlled by eight different enzymes. As this production process of heme progresses, several different intermediate compounds (heme precursors) are created and modified. If one of the essential enzymes in heme production is deficient, certain precursors may accumulate in tissues (especially in the bone marrow or liver), appear in excess in the blood, and get excreted in the urine or stool. The specific precursors that accumulate depend on which enzyme is deficient. Porphyria results in a deficiency or inactivity of a specific enzyme in the heme production process, with resulting accumulation of heme precursors.
How is porphyria diagnosed?
Porphyria is diagnosed through blood, urine, and stool tests, especially at or near the time of symptoms. Diagnosis may be difficult because the range of symptoms is common to many disorders and interpretation of the tests may be complex. A large number of tests are available, however, but results among laboratories are not always reliable.
How is porphyria treated?
Each form of porphyria is treated differently. Treatment may involve treating with heme, giving medicines to relieve the symptoms, or drawing blood. People who have severe attacks may need to be hospitalized.
What are the signs and symptoms of porphyria?
The signs and symptoms of porphyria vary among types. Some types of porphyria (called cutaneous porphyria) cause the skin to become overly sensitive to sunlight. Areas of the skin exposed to the sun develop redness, blistering and often scarring.
The symptoms of other types of porphyria (called acute porphyrias) affect the nervous system. These symptoms include chest and abdominal pain, emotional and mental disorders, seizures and muscle weakness. These symptoms often appear quickly and last from days to weeks. Some porphyrias have a combination of acute symptoms and symptoms that affect the skin.
Environmental factors can trigger the signs and symptoms of porphyria. These include:
Alcohol
Smoking
Certain drugs, hormones
Exposure to sunlight
Stress
Dieting and fasting
What do we know about porphyria and heredity?
Most of the porphyrias are inherited conditions. The genes for all the enzymes in the heme pathway have been identified. Some forms of porphyria result from inheriting one altered gene from one parent (autosomal dominant). Other forms result from inheriting two altered genes, one from each parent (autosomal recessive). Each type of porphyria carries a different risk that individuals in an affected family will have the disease or transmit it to their children.
Porphyria cutanea tarda (PCT) is a type of porphyria that is most often not inherited. Eighty percent of individuals with PCT have an acquired disease that becomes active when factors such as iron, alcohol, hepatitis C virus (HCV), HIV, estrogens (such as those used in oral contraceptives and prostate cancer treatment), and possibly smoking, combine to cause an enzyme deficiency in the liver. Hemochromatosis, an iron overload disorder, can also predispose individuals to PCT. Twenty percent of individuals with PCT have an inherited form of the disease. Many individuals with the inherited form of PCT never develop symptoms.
If you or someone you know has porphyria, we recommend that you contact a genetics clinic to discuss this information with a genetics professional. To find a genetics clinic near you, contact your primary doctor for a referral.
What triggers a porphyria attack?
Porphyria can be triggered by drugs (barbiturates, tranquilizers, birth control pills, sedatives), chemicals, fasting, smoking, drinking alcohol, infections, emotional and physical stress, menstrual hormones, and exposure to the sun. Attacks of porphyria can develop over hours or days and last for days or weeks.
How is porphyria classified?
The porphyrias have several different classification systems. The most accurate classification is by the specific enzyme deficiency. Another classification system distinguishes porphyrias that cause neurologic symptoms (acute porphyrias) from those that cause photosensitivity (cutaneous porphyrias). A third classification system is based on whether the excess precursors originate primarily in the liver (hepatic porphyrias) or primarily in the bone marrow (erythropoietic porphyrias). Some porphyrias are classified as more than one of these categories.
What are the cutaneous porphyrias?
The cutaneous porphyrias affect the skin. People with cutaneous porphyria develop blisters, itching, and swelling of their skin when it is exposed to sunlight. The cutaneous porphyrias include the following types:
Also called congenital porphyria. This is a rare disorder that mainly affects the skin. It results from low levels of the enzyme responsible for the fourth step in heme production. It is inherited in an autosomal recessive pattern.
An uncommon disorder that mainly affects the skin. It results from reduced levels of the enzyme responsible for the eighth and final step in heme production. The inheritance of this condition is not fully understood. Most cases are probably inherited in an autosomal dominant pattern, however, it shows autosomal recessive inheritance in a small number of families.
A rare disorder that mainly affects the skin. It results from very low levels of the enzyme responsible for the fifth step in heme production. It is inherited in an autosomal recessive pattern.
A rare disorder that can have symptoms of acute porphyria and symptoms that affect the skin. It results from low levels of the enzyme responsible for the sixth step in heme production. It is inherited in an autosomal dominant pattern.
The most common type of porphyria. It occurs in an estimated 1 in 25,000 people, including both inherited and sporadic (noninherited) cases. An estimated 80 percent of porphyria cutanea tarda cases are sporadic. It results from low levels of the enzyme responsible for the fifth step in heme production. When this condition is inherited, it occurs in an autosomal dominant pattern.
A disorder that can have symptoms of acute porphyria and symptoms that affect the skin. It results from low levels of the enzyme responsible for the seventh step in heme production. It is inherited in an autosomal dominant pattern.
What are the acute porphyrias?
The acute porphyrias affect the nervous system. Symptoms of acute porphyria include pain in the chest, abdomen, limbs, or back; muscle numbness, tingling, paralysis, or cramping; vomiting; constipation; and personality changes or mental disorders. These symptoms appear intermittently. The acute porphyrias include the following types:
This is probably the most common porphyria with acute (severe but usually not long-lasting) symptoms. It results from low levels of the enzyme responsible for the third step in heme production. It is inherited in an autosomal dominant pattern.
A very rare disorder that results from low levels of the enzyme responsible for the second step in heme production. It is inherited in an autosomal recessive pattern.
What is δ-Aminolevulinic Acid Dehydratase Porphyria (ADP)?
Friday - April 12, 2019 @ 07:00:04
What is δ-Aminolevulinic Acid Dehydratase Porphyria (ADP)?
ADP is more severe than the other acute porphyrias and can present in childhood. It is an inherited genetic condition, but is extremely rare. Only ~10 cases have been reported worldwide and all of the reported cases have been males, in contrast to the other acute porphyrias where more women have symptoms. In ADP, the gene responsible is ALAD which produces the enzyme δ-aminolevulinic acid dehydratase. When this enzyme is working properly, porphyrins build up and can cause symptoms similar to those seen in AIP.
How is δ-Aminolevulinic Acid Dehydratase Porphyria diagnosed?
Biochemical testing means looking for biomarkersin the blood or urine. To diagnose ADP, measurements of porphobilinogen (PBG), aminolevulinic acid (ALA), and total porphyrins in the urine should be done. Also porphyrins in the blood should be measured. The level of PBG in the body can vary so the best time to take samples is during an acute attack (e.g. when someone is having abdominal pain, etc). Slight elevations in porphyrins are not diagnostic of ADP; the levels need to be very high.
What are treatments for δ-Aminolevulinic Acid Dehydratase Porphyria?
The treatments and preventive measures are the same as in AIP.
How is δ-Aminolevulinic Acid Dehydratase Porphyria Inherited?
ADP is an autosomal recessive condition. Autosomal means that the defect is not on the chromosomes that determine sex, and recessive means that both copies of the gene are mutated. The gene that causes ADP is called ALAD.
MEET MILTON CUBAS, APF Member and ALAD Porphyria (ADP) patient. My name is Milton Eduardo Cubas and I am 29 years old. Sports and I physical activities with others are two of my favorite things. My current studies at Miami Dade College are to become a math teacher. I had my first porphyria attack in a very hot summer day of 2000 at a castle in Cartagena, Colombia named Castillo San Felipe de Barajas. I was with family members and I remember feeling fatigued while taking a tour of the castle. I drank a whole bottle of water after leaving. Once I got in a taxi I felt nausea. I felt tired. We stopped to eat something but I would vomit water until nothing would come out. I had gone to hospitals in Colombia and they couldnt find anything. I remember feeling a lot of pain while looking for anyone to tell me what is going on. I had to cut my trip to Colombia short because we couldnt find anyone to help me and my pain was getting worse. I went to Miami Childrens Hospital and my attack got better and the doctors still couldnt find anything. I would have an attack every month or two for a year until a doctor named Elsa Vasconsuelos told me that I may have Porphyria. Soon after that, my dad found Dr. Anderson. I would take two or three trips to Galveston, Texas until he diagnosed me in February 2003 with aminolevulinic acid dehydratase-deficient porphyria, which is abbreviated as ADP. Through my teenage years, I had attacks nearly every month. Once I hit my 20s the attacks spread out more. The longest Ive spent without an attack is two and a half years but after that I had 3 attacks really close to each other that affected my neuropathy a lot. My last attack was mid-July of 2017. I get hematin every single Friday. I have tried spacing out the hematin to every 10 days or every 14 days but I would get an attack after that. During the attack, I would have to be hospitalized to get hematin for four days every day. I get heme in an infusion center. Some of my negative experiences are that some nurses dont know how to administer the hematin the correct way when Im hospitalized. The neuropathy has affected me every day from dressing myself to doing homework. I have trouble with extending my fingers that stops me from doing hand gestures. I have foot drop where I could lose my balance easily. It has forced me to try find anywhere where there is AC. I could last longer in the sun now but in the summer I cant. There have been positive experiences, too. I have met a lot of wonderful people, nurses and doctors, who have helped me when Im trying to get better. My Family has supported me and helped me whenever I couldnt drive or when I was hospitalized. I want to become a Math teacher and I am studying really hard to graduate. I want to get my hands better. For the people who have Porphyria: Knowledge is Power. The more you know about Porphyria the stronger you will be. It is also important to participate in research and I am in the longitudinal study at UTMB.
What is Variegate Porphyria (VP)?
Thursday - April 11, 2019 @ 07:00:04
What is Variegate Porphyria (VP)?
VP is an inherited genetic condition with similar clinical signs and symptoms as AIP, but is more rare than AIP. VP is especially common in South African individuals of Dutch ancestry, where it has been estimated that 3 in 1,000 of the Caucasian population is affected. In VP, the gene responsible is PPOX which produces the enzyme coproporphyinogen oxidase, and without this enzyme working properly porphyrins build up and can cause symptoms similar to those seen in AIP. However the acute attacks can be milder in people with VP when compared to AIP. Patients with VP have the same slightly increased risk of liver cancer that AIP patients have. Unlike AIP, people with VP can also have blistering on their skin in response to sun exposure and the primary affected areas are on the back of the hands and face. The occurrence of blistering skin lesions are much more common in VP than in HCP and are not easily treated. The only effective preventive measure is use of protective clothing and avoidance of prolonged sun exposure. As in AIP, about 80-90% of patients with VP mutations will not develop symptoms.
How is Variegate Porphyria diagnosed?
There are two types of testing; biochemical, meaning looking for biomarkersin the blood or urine, and genetic, meaning looking at the gene we know causes the disease directly from a blood sample.
Biochemical: To diagnose VP, measurement of porphobilinogen (PBG), aminolevulinic acid (ALA), and total porphyrins in the urine should be done. Also porphyrins in the blood should be measured. The level of PBG in the body can vary so the best time to take samples is during an acute attack (e.g. when someone is having abdominal pain, etc.). Slight elevations in porphyrins are not diagnostic of VP; the levels need to be very high.
Genetic: A blood sample is used to look at a persons genes and by doing this it is possible to see if their genes have changes that can cause disease, called mutations. VP is caused by mutations in the PPOX gene. Genetic testing is recommended for patients even if they have a biochemical diagnosis of VP.
If a patient has a mutation, their immediate family members should be tested for that same mutation as well. This includes their parents, their siblings, and any children they may have. This will allow all family members to receive appropriate care and counseling even though 80-90% of people with a mutation will not have symptoms of VP.
What are treatments for Variegate Porphyria?
The treatments and preventive measures are the same as in AIP. In addition patients with blistering from sun exposure will need to protect themselves from sunlight by using sun protective clothing and avoiding prolonged sun exposure.
How is Variegate Porphyria Inherited?
VP is an autosomal dominant condition. Autosomal means that the defect is not on the chromosomes that determine sex, and dominant means that you only need to inherit one mutated gene to manifest the disease. The gene that causes HCP is called PPOX.
Genes are inherited randomly, so a parent has an equal chance of passing on either copies of each gene. Since most VP patients have one mutated copy and one normal copy, this means that each of their children will have a 50% chance of inheriting the mutated copy and 50% chance of inheriting the working copy.
My story is probably not much different than most. I was misdiagnosed most of my life, sent to psychologists because physicians just couldn't find a cause for my illness. Finally, after an incorrect Lupus diagnosis and a week of testing, I received my Variegate Porphyria diagnosis.
As a child growing up in Tampa, FL, I would constantly complain that the sun hurt my skin and my eyes but "children" were not supposed to wear sun glasses. Today I am rarely seen without them, day or night. I craved sweets and of course "children" should not eat too much sugar! I went to a boarding school most of my life and "they" had the same rules about sun protection and sweets the same as my parents. For reasons like this, I have always wanted to write a book "If you Can't See itIt Must Not Be Real" for all of "us" who are ill but it can't be seen, and so it must be in our "heads"!!
How might I be different? I have always known I am a survivor in life and have tried to maintain a positive attitude. At a very early age, I chose the "high road". I have pushed myself both mentally and physically to go beyond and not make my disease a focus in my life. I worked hard to receive a good education, eventually owned a successful cooperation, raised three step children and married twice. Now I ride and show horses (jumpers), work out daily and volunteer whenever I can (I have recently been helping women coming out of prison to assimilate back into the business world) and continue to look for areas and people to add to my life.
I have not had the best experiences with physicians. I was made worse by a physician claiming I would NEVER meet anyone else with my disease. He was like many physicians, who know very little about the disease and read only one chapter in medical school. Nonetheless, he was a self-proclaimed expert on porphyria with his own "treatments." I am alive today after surviving his dilation toxicity for three years and having a gland removed by his surgeon friend. This left me with permanent double vision at three and half feet out. Thus, reading is almost impossible for me other than my large computer screen.
Next, I broke most of my fingers and had another serious problem. I am going through implant surgery now because of mishandling by a dentist. Then I was left bleeding to death in "after surgery" care, overdosed with morphine, followed by a surgeon who was drunk and performed ovarian surgery incorrectly. I could go on, but I suffice to write this to let you know "we" must be our own advocates in health care. I still seek the good in all I meet and do believe there are wonderfully talented and gifted physicians, but my experiences have certainly caused me to be vigilant over my own care. As for porphyria, I thank God for Dr. Anderson, his colleagues and Desiree.
My attitude is to count my blessings, allow myself some few "moments" of self pitypick myself up and move forward. We all have challenges in life and sometimes hurt so badly that it is hard to live another day--but we do. We need to share information and stories, support to each other and always keep our sense of humor. I try to look at what I have rather than what I don't have.
In the end it will always be our "attitudes" that drive our lives. As a very wise man long ago told me, "You have two choices in the morning when you start your day" for whatever reason, hearing him tell me on that day changed my life, and I say this to all of you hoping I can make the same difference. Good luck and God Bless!
My name is Abdul Waheed Butt. I lived in Pakistan I am facing skin problem name Congenital Erythropoietic Porphyria (Gunther Disease) CEP. When I was born, I was completely normal like other children. When I was 2-months-old, my mother cut my hand nails there are starting water from it. When the nails water touch my face it becomes Water Blisters on my whole face. My parents checked me from so many Doctors in Pakistan, but they have no idea or treatment how to solve that problem. At-last, we found a skin specialist Dr. Ishfaq Ahmed who said that the disease name is CEP. He advised me to avoid sunlight and covered when I go out. The only treatment of this disease is to avoid sunlight. Then my parents covered me all the time for my safety as they can.
When I was 7-years-old the effect of the disease is gradually decreased and the Blisters which are appeared automatically are disappeared. But there are so many scars on my face and hands.
After that I am starting my study and completed my Graduation degree in Commerce. During my study I have facing so many problems from classmates, when they see me, they behave like that I am not a human or came from other planet.
After graduation I have done my computer course and using internet, I find skin Specialist Doctors in all over the world and send them my pictures and story to review my case. Approximately five years of regular struggle I have received a call of Miss Desiree Lyon from America. She tells me about the details of the disease and advise me to avoid sunlight as you can. Then my Story and pic is displayed on APF website and add my name in Facebook APF Group. In this group I make lots of friends who have CEP and EPP.
APF send me a doctor kit for my blood and urine test to clarify that my disease name after test the report result is CEP.
Now my age is 30 and I am working as Computer Clerk in Medical College in Pakistan. I have so many friends in Facebook it is so useful for all CEP members for discussed their problem and get a fair advised from each others. I will be sure that the treatment of the disease is discovered by the help of God and we all get the treatment soon.
God said that There is no disease in the Earth without Cure
And last, I would like to thank again the APF, Desiree Lyon and Amy Chapman who help me. Thank you so much.
Abdul Waheed Butt House no CB 408, St no 16, Jhanda Chichi Rawalpindi, Pakistan 00923325458654 waheed_butt98@yahoo.com
Congenital erythropoietic porphyria (CEP) is the rarest type of porphyria and is commonly seen in infancy.[1] It is characterized by severe skin photosensitivity that may lead to scarring, blistering, and increased hair growth at the face and back of the hands.[2][3] Photosensitivity and infection may cause the loss of fingers and facial features.[1] Symptoms of CEP range from mild to severe and may include excessive hair growth throughout the body (hypertrichosis), reddish discoloration of the teeth, anemia, and reddish-colored urine.[4] In CEP, there is a defect in the synthesis of heme within the red blood cells of bone marrow.[3][4] This defect leads to an increase in the buildup and, therefore, waste of porphyrin and its precursors, which leads to the signs and symptoms.[3] Inheritance is autosomal recessive. It is caused by mutations in the UROS gene.[3] Treatment for CEP may include a bone marrow transplant and hematopoietic stem cell cord blood transplantation.[2][1]Blood transfusions or spleen removal may also reduce the amount of porphyrin produced by the bone marrow. Affected people must avoid sunlight exposure.[1]
This table lists symptoms that people with this disease may have. For most diseases, symptoms will vary from person to person. People with the same disease may not have all the symptoms listed. This information comes from a database called the Human Phenotype Ontology (HPO) . The HPO collects information on symptoms that have been described in medical resources. The HPO is updated regularly. Use the HPO ID to access more in-depth information about a symptom.
The resources below provide information about treatment options for this condition. If you have questions about which treatment is right for you, talk to your healthcare professional.
Management Guidelines
The American Porphyria Foundation offers a document that includes information about porphyria, types, testing, and treatment with Panhematin. Click the "document" link above to view these guidelines.
If you need medical advice, you can look for doctors or other healthcare professionals who have experience with this disease. You may find these specialists through advocacy organizations, clinical trials, or articles published in medical journals. You may also want to contact a university or tertiary medical center in your area, because these centers tend to see more complex cases and have the latest technology and treatments.
If you cant find a specialist in your local area, try contacting national or international specialists. They may be able to refer you to someone they know through conferences or research efforts. Some specialists may be willing to consult with you or your local doctors over the phone or by email if you can't travel to them for care.
You can find more tips in our guide, How to Find a Disease Specialist. We also encourage you to explore the rest of this page to find resources that can help you find specialists.
Related diseases are conditions that have similar signs and symptoms. A health care provider may consider these conditions in the table below when making a diagnosis. Please note that the table may not include all the possible conditions related to this disease.
Conditions with similar signs and symptoms from Orphanet
Differential diagnosis can include hepatoerythropioetic porphyria (see this term).
Research helps us better understand diseases and can lead to advances in diagnosis and treatment. This section provides resources to help you learn about medical research and ways to get involved.
Clinical Research Resources
ClinicalTrials.gov lists trials that are related to Congenital erythropoietic porphyria. Click on the link to go to ClinicalTrials.gov to read descriptions of these studies.
Please note: Studies listed on the ClinicalTrials.gov website are listed for informational purposes only; being listed does not reflect an endorsement by GARD or the NIH. We strongly recommend that you talk with a trusted healthcare provider before choosing to participate in any clinical study.
Patient Registry
The Porphyrias Consortium is a team of doctors, nurses, research coordinators, and research labs throughout the U.S., working together to improve the lives of people with this condition through research. The Porphyrias Consortium has a registry for patients who wish to be contacted about clinical research opportunities.
Support and advocacy groups can help you connect with other patients and families, and they can provide valuable services. Many develop patient-centered information and are the driving force behind research for better treatments and possible cures. They can direct you to research, resources, and services. Many organizations also have experts who serve as medical advisors or provide lists of doctors/clinics. Visit the groups website or contact them to learn about the services they offer. Inclusion on this list is not an endorsement by GARD.
RareConnect has an online community for patients and families with this condition so they can connect with others and share their experiences living with a rare disease. The project is a joint collaboration between EURORDIS (European Rare Disease Organisation) and NORD (National Organization for Rare Disorders).
Living with a genetic or rare disease can impact the daily lives of patients and families. These resources can help families navigate various aspects of living with a rare disease.
Financial Resources
The HealthWell Foundation provides financial assistance for underinsured patients living with chronic and life-altering conditions. They offer help with drug copayments, deductibles, and health insurance premiums for patients with specific diseases. The disease fund status can change over time, so you may need to check back if funds are not currently available.
These resources provide more information about this condition or associated symptoms. The in-depth resources contain medical and scientific language that may be hard to understand. You may want to review these resources with a medical professional.
Where to Start
Genetics Home Reference (GHR) contains information on Congenital erythropoietic porphyria. This website is maintained by the National Library of Medicine.
The National Human Genome Research Institute's (NHGRI) website has an information page on this topic. NHGRI is part of the National Institutes of Health and supports research on the structure and function of the human genome and its role in health and disease.
The National Organization for Rare Disorders (NORD) has a report for patients and families about this condition. NORD is a patient advocacy organization for individuals with rare diseases and the organizations that serve them.
In-Depth Information
Medscape Reference provides information on this topic. You may need to register to view the medical textbook, but registration is free.
Online Mendelian Inheritance in Man (OMIM) is a catalog of human genes and genetic disorders. Each entry has a summary of related medical articles. It is meant for health care professionals and researchers. OMIM is maintained by Johns Hopkins University School of Medicine.
Orphanet is a European reference portal for information on rare diseases and orphan drugs. Access to this database is free of charge.
PubMed is a searchable database of medical literature and lists journal articles that discuss Congenital erythropoietic porphyria. Click on the link to view a sample search on this topic.
Questions sent to GARD may be posted here if the information could be helpful to others. We remove all identifying information when posting a question to protect your privacy. If you do not want your question posted, please let us know.
This all began August 2017 after starting a new job. I started having severe, crampy, menstrual like abdominal pain that came and went seemingly at random. After a couple of months of this, and multiple Dr. and ER visits, my mother suggested I take an Ibuprophen to get through the work day. Wow!, was that a mistake! Within 10 minutes of taking it my pain was 100x worse and visibly swollen. From that moment on the pain was constant and increasing in severity, causing me to go the ER again. The Drs acted like I was a hypochondriac drug addict and sent me home after running routine tests, that came back normal. I was referred to a GI specialist who thought I had stomach issues and ordered an endoscopy(that was of course normal). After hearing my results I was confused and defeated, and I was beginning to wonder if I was being a hypochondriac. But I knew that what I saw and felt in my body was not normal. Around the beginning of December, my mother called and suggested I try the porphyria diet. So I did, and within 2 weeks I was starting to feel better.
Fast forward to April 2018, I had my first appointment with Dr. Silver, a porphyria specialist. Due to the fact that I vomited the entire 3 hour appointment, and I had a family history of AIP; Dr Silver said he thought I had AIP. I continued to vomit for the next 6 days, with each day worse than the last so I went to the ER on the 6th day.
Dr. Silver oversaw my treatment and Hemin infusions my entire stay. I thought that was it, my attack was over, but boy was I wrong. I had attacks increasing in severity every 2 weeks for the next few months before I saw they were hormone induced. I then began the long process of getting Lupron injections to stop my hormone production. While waiting for Lupron, I had the worst attack yet, this one effected my legs. The pain was so severe I couldnt move my legs. This happened after I had an attack on the nerves that control my muscles, which left me on the floor unable to get up one morning. I received my first Lupron shot during the attack on my legs, and I also received 10 bags of Hemin during my 14 day stay. The Lupron took 2 months to fully suppress my hormones, and during that waiting period I my worst attack to date. Due to the pain, I was unable to move my own body and had to be moved by others. I also was dealing with the full body swelling that had always been going on but had gotten infinitely worse during this attack.
The inflammation has not gone down since that attack. I am also part of a drug study and have been for months. This drug seems to be helping me recover and end the attack. I am in a wheelchair and have been since the nerves that control the muscles were effected. But I am improving and walking some, and with the support of my family I will defeat AIP!
Severe abdominal pain: the most common AIP symptom
The most common symptom of AIP is severe abdominal pain that usually cannot be relieved with pain medicine such as Advil (ibuprofen) or Tylenol (acetaminophen). More than 85% of people who develop AIP symptoms have abdominal pain.
Experiencing symptoms is known as having an AIP attack.Symptoms may occur for a set period of time, then go away only to come back later.
Common AIP symptoms
Symptoms can occur in many different areas of your body during an AIP attack. These include:
GASTROINTESTINAL (GI) SYMPTOMS
Abdominal pain
Vomiting
Constipation
Diarrhea
GASTROINTESTINAL (GI)
URINARY SYSTEM
BRAIN OR NERVOUS SYSTEM
HEART OR BLOOD VESSELS
Early diagnosis and treatment of AIP are critical
AIP attacks can be very serious. And symptoms may get worse over time. Untreated attacks can cause serious damage to your nervous system including paralysis, and even death. That's why early diagnosis and treatment of AIP is so important.
If you have any of the symptoms listed above, talk to your doctor right away.
What triggers AIP attacks?
Steroid hormones, particularly estrogen and progesterone. These hormones fluctuate the most during the 2 weeks before a womans menstrual periods start.
Unhealthy behaviors such as drinking alcohol, smoking, or using illegal drugs.
Stress on the body caused by infections, surgery, or physical exhaustion.
Certain prescription drugs. Attacks can also be triggered by starting a new prescription drug.
Changes in eating patterns such as fasting or crash dieting.
AIP is caused by a partial lack of an enzyme known as porphobilinogen deaminase (PBGD). An enzyme is a type of protein that helps to regulate the bodys tissues and organs. Enzymes carry out almost all of the thousands of chemical reactions that take place in cells.
If you have AIP, you have about half of the normal amount of PBGD in your body. This is usually enough for your body to do what it is supposed to do. But triggers like those listed above can upset your body's chemical balance enough to cause symptoms.
Important Safety Information PANHEMATIN should only be used by doctors experienced in the management of porphyrias in hospitals where the recommended clinical and laboratory diagnostic and monitoring practices are available.
TAKE STEPS TO HELP MANAGE YOUR AIP:
Try to identify your possible triggers. Then try to reduce or avoid as many as you can. If you continue to experience attacks, keep writing down suspected triggers. Look for patterns of things that occurred right before an attack to identify any changes you can make. Talk to your doctor if you need help.
Be careful when changing your eating patterns. If you want to lose weight, get advice from your doctor or nutritionist before starting any diet.
Call your doctor at the first sign of an attack. Repeat attacks are often similar. They may start the same way and you may have the same symptoms.
Wear a medical alert bracelet. Doctors need to know about your AIP so that they do not prescribe drugs that may make your AIP worse.
Indications And Usage
PANHEMATIN is a hemin for injection prescription medication used to relieve repeated attacks of acute intermittent porphyria related to the menstrual cycle in affected women, after initial carbohydrate therapy is known or suspected to be inadequate.
Limitations of Use
Before giving PANHEMATIN, consider an appropriate trial of carbohydrates [i.e., 400 grams of glucose (a carbohydrate or sugar) per day for 1 to 2 days].
Attacks of porphyria may progress to a point where irreversible nerve damage has occurred. PANHEMATIN therapy is intended to prevent an attack from reaching the critical stage of nerve breakdown. PANHEMATIN is not effective in repairing nerve damage that has already occurred.
Important Safety Information
PANHEMATIN is not for patients known to be allergic to this drug.
Vein inflammation is possible. Use a large arm vein or a central line to administer PANHEMATIN to minimize the possibility of vein inflammation.
Elevated iron levels may occur. Your doctor must monitor your iron levels if you receive multiple courses of PANHEMATIN.
PANHEMATIN has a passing and mild blood thinning effect. Avoid blood thinners during PANHEMATIN therapy.
Reversible kidney shutdown has occurred when too high a dose of PANHEMATIN was given in a single dose.
PANHEMATIN may carry a risk of transmitting agents that can cause infections, such as viruses, and theoretically, the Creutzfeldt-Jakob disease (CJD) agent.
Most common side effects are headache, fever, infusion site reactions, and vein inflammation.
To report SUSPECTED SIDE EFFECTS, contact Recordati Rare Diseases Inc. at 1-888-575-8344, or FDA at 1800-FDA-1088 or www.fda.gov/medwatch.
When taking PANHEMATIN, do not take drugs such as estrogens (e.g., oral contraceptives), barbiturates (drugs that help with sleep and used to treat epilepsy) or steroids (body hormone-like drugs), because such drugs can trigger an attack or make an attack worse.
My name is Cassandra King. There are three main things that define me: I am the daughter of the King of Kings (saved by grace through Jesus), I am the wife of Russ King, and I am the mama of Joshua King. There are, of course, other hats I wear: Im a daughter, a sister, an auntie, a friend, a volunteer, etc. And though, I really loathe to admit it, there is one other thing that defines me pain. Constant, unrelenting, heart-breaking pain. A broken body.
I lived the first couple decades of my life with excellent health, perfect attendance in school, no limitations physically. When Russ and I married, we had no sense of concern for anything health wise, and we didnt actually invest in health insurance for ourselves until wed been married for over 2 years. I was a work until the project is finishedkinda gal, eat when its convenientkinda gal, and rest when everythings completedkinda gal. Russ and I married in May of 2004. After spending a couple months working the summer hay season with his folks, we made our home in southern California so Russ could continue in Bible college. I worked full-time and Russ went to school. We returned to Klamath Falls in 2005 for Russ to continue farming with his folks. And I got a job at our church as the secretary.
In the first week of December of 2006, I had a crazy, full work week of events, meetings, hosting, planning, and office work. I went to church on Sunday morning feeling tired and run down a bit, but knew I had Mondays off, so Id be able to catch up on sleep. This is not what happened at all. I started vomiting, and we initially thought Id just caught a bug from being over tired the past week. It got worse on Tuesday, and even worse on Wednesday. Wednesday night we spent in the ER, sent home with a diagnosis of a very large ovarian cyst, pain medication and nausea medication. Thursday was worse, nothing was staying in, so no help from the medications, I started shaking/seizing, seeing things, and more. We went into the ER again Thursday night, and didnt leave the hospital again for almost 4 weeks. I remember snippets of this time, my Russ has to remember it all. I was in so much pain, originating in my abdomen and spreading through my whole body. I couldnt eat, couldnt keep anything down. I was extremely weak, couldnt walk, couldnt dress myself or care for myself. Every touch hurt, everything was fire. They did every test in the playbook, trying to find out what was wrong. All the tests were coming back fairly normal, but I was getting sicker and weaker and worse. Finally, they decided to put in a central line and give me tpn (total protein nutrition) to save my life. I had lost 30+ pounds, was under 100 pounds. After coming and saying that I was the enigma of the hospital, that they couldnt figure out what was wrong with me and couldnt stop it; the doctors ordered a final test. On Christmas Eve, one of the doctors came in wearing a Santa hat and said, were going to send you home for Christmas. I didnt know they had also told Russ that they couldnt guarantee Id make it, to try to keep her as comfortable as possible. We left with no answers. I was only 22 years old.
I spent the next 3 months recovering, not returning to work until early March. The recovery was hard, I knew how to walk, talk, eat, care for myselfâ?¦. But had I no strength or stamina to be able to do any of it for myself. A month after leaving the hospital, we finally had an answer. That final test in the hospital came back with levels all relatively normal, except for one level the porphobilinogen (PBG) level. It was 300 times what it should be. Gods providence allowed that one of the residents at the clinic to be the one to see us that day, and she happened to recall a crazy story from med school about vampires and porphyria, and she connected it with my test results. When she first said this, I remember thinking, this is nuts, and I am going to die in this town with home-town medicine. But she went on to say that this disease, Porphyria, has various kinds. And they thought I may have type called Acute Intermittent Porphyria.
We were sent up to OHSU to the doctor who had actually written the emedicine.com paper on this disease. He looked at my test results and asked a few questions. Then he told us, Theres 5 porphyria specialists in the country. Im the one for the northwest part, and youre the first case Ive seen of AIP in 10 years. This disease is SUPER RARE! Most doctors and nurses have never even heard of it before. It was Gods grace to allow that resident to be the one to see me that day and for her to remember the story about vampires - Because it gave me a diagnosis the very first time, I got sick. Im so grateful. The specialist went on to say that 60-80% of people that have 1 attack of AIP will never have another, so I could go and live my life normally. We thought, if you have to have a disease, this is the one to have!
So, after I had recovered my strength and abilities, I returned to my normal life work at the church, farm wife, everything. Six months later, we came to understand, I am in the 20-40% of people who will have more and more attacks. In September 2007, I spent a week and a half in the hospital, treating the attack with pain management, nausea medication and hematin treatments through a central line. I was home for 5 days, then back in to the hospital for another 6 days of pain & nausea meds and hematin in yet another central line. Followed by another month and a half of recovery at home. This began a cycle of every 3 months having a severe AIP attack from October 2007-October 2010. Some could be managed at home with pain & nausea meds orally, lots of rest and diet. Some required hospitalization for iv pain and nausea meds, a central line for numerous hematin treatments. The attacks got harder, more severe, with unbelievable pain, and came with new scary symptoms: seizures, bouts of hysteria and anxiety, confusion and mind/memory fog, neuropathy in my hands & arms, feet & legs, motor neuropathy (partial paralysis), and more horrors.
Now, Im kinda stubborn and Id try to outrun these attacks. But once the motor neuropathy would begin, Id cave quickly to going into the hospital for treatment. Russ became the pastor of our church in 2008, and I had the blessing of continuing as the church secretary for him for 2 more years. But, as I was still having these cyclical attacks, it left Russ and his staff under further pressure when I was out for health because they had to do my job as well as their own. And after 3 years of all these attacks, my body was not bouncing back after the attacks. My body was permanently damaged. So, I had to come to the hard decision to medically retirequit my job, that I loved deeply. I had no child at home, I was 26 years old, and I was unable to work because of my health.
In August of 2010, we went for our yearly checkup appt with our porphyria specialist at OHSU. We told him how the pain was not going away after this most recent attack, the neuropathy wasnt going away. What do we do?!? And this is when my life really changed from porphyria. He told us that unfortunately it was time for me to get a chest port and to do hematin treatments as preventative measures monthly. Two months later, in October 2010, I had another severe attack requiring hospitalization. I had my port surgery completed during this attack for the hematin treatments to be administered through the port. This began monthly hematin treatments with the goal of preventing severe AIP attacks, allowing my body the chance to heal the damage and not create more with further attacks. For the first 4 years, I did treatments once a month. For the last 4 years, I have a treatment every other week. Weve been able to stop the 3-month cyclical attacks, but theres still been 4 severe attacks in these years requiring hospital intervention and weeks of recovery. The most recent one being the beginning November 2018.
The treatments have had some success in preventing the number of attacks, but not much success in letting my body heal all the damage incurred from AIP. Theres not been 1 day in the past 9 years without severe, constant, intense pain and unrelenting, agonizing neuropathy in my hands and feet. Where we once thought porphyria will not be something we even think or worry about, is now replaced with the reality of how porphyria affects every hour and every day of my life. It affects not only my body and my everyday life, but also my husband and our marriage, our son, our family, our church, everything is severely impacted by AIP. My body is broken.
But one thing about porphyria, or rather the form I have AIP, is that its a relatively invisible illness the reality of the horrors are not easily seen in my body (makeup and a curling iron do a lot. Each morning I wake up in incredible, massive pain physical and neurological. And then I try to figure out what my body will allow for the day in terms of energy, strength and pain endurance. Yes, my body is broken, and my physical state is very hard every single day. But God can take whats broken and so damaged, and make it beautiful. The beauty of brokenness is that God can put the pieces back together. Is there beauty in every brokenness? It dependsâ?¦ it becomes beautiful when it makes one turn to God. (Paul Jan Chi) He is my hope, He is my sustainer. God is sovereign and Jesus is enough. God is the reason my broken body bears a smile every day He gives perfect peace and immeasurable joy in the midst of the hard, broken, pain-filled days. His loves in amazing and gives me strength. My prayer each day is to trust God in all things, to obey God and to glorify God through my thoughts, words and actions.
It is all for HIS glory!
Blessed be the God and Father of our Lord Jesus Christ, who according to His abundant mercy has begotten us again to a living hope through the resurrection of Jesus Christ from the dead, to an inheritance incorruptible and undefiled and that does not fade away, reserved in heaven for you, who are kept by the power of God through faith for salvation ready to be revealed in the last time. In this you greatly rejoice, though now for a little while, if need be, you have been grieved by various trials, that the genuineness of your faith, being much more precious than gold that perishes, though it is tested by fire, may be found to praise, honor, and glory at the revelation of Jesus Christ, whom having not seen you love. Though now you do not see Him, yet believing, you rejoice with joy inexpressible and full of glory, receiving the end of your faiththe salvation of your souls. (1 Peter 1:3-9)
Updated info on PCT PubMed
Monday - April 8, 2019 @ 07:00:06
Mol Genet Metab. 2019 Jan 18. pii: S1096-7192(18)30579-1. doi: 10.1016/j.ymgme.2019.01.004. [Epub ahead of print]
Porphyria cutanea tarda (PCT) is the most common human porphyria, due to hepatic deficiency of uroporphyrinogen decarboxylase (UROD), which is acquired in the presence of iron overload and various susceptibility factors, such as alcohol abuse, smoking, hepatitis C virus (HCV) infection, HIV infection, iron overload with HFE gene mutations, use of estrogens, and UROD mutation. Patients with familial or type II PCT due to autosomal dominant UROD mutation also require other susceptibility factors, as the disease phenotype requires hepatic UROD deficiency to below 20% of normal. PCT clinically manifests with increased skin fragility and blistering skin lesions on sun exposed areas. The common age of presentation is 5th to 6th decade and occurs slightly more commonly in males. Although mild liver biochemical profile are common, advanced fibrosis and cirrhosis with hepatocellular carcinoma (HCC) can occasionally develop. Screening for HCC using ultrasound examination is recommended in PCT patients, especially with cirrhosis and advanced fibrosis. PCT is effectively and readily treatable with the use of either repeated phlebotomy or use of 100â?¯mg hydroxychloroquine orally twice a week, and both the treatments are equally effective and safe. With the advent of new or direct antiviral agents for HCV infection, treatment of concomitant HCV has become safer and effective. Data are emerging on the benefit of these drugs as monotherapy for both PCT and HCV. After the achievement of remission of PCT, there remains a potential for relapse, especially when the susceptibility factors are not adequately controlled. Scanty data from retrospective and observational studies shows the relapse rate to be somewhat higher after remission with low-dose hydroxychloroquine as compared to phlebotomy induced remission. Future studies are needed on exploring mechanism of action of 4-aminoquinolines, understanding interaction of HCV and PCT, and relapse of PCT on long-term follow-up.
I consider myself to be a pretty confident, healthy, and strong woman with a whole lot of life to live. Life is beautiful and I never knew how much I took it for granted until 2016 hit me. I didnt feel like myself at all. I was the heaviest I had ever been and couldnt muster up the energy to get through the day. Juggling a full-time job along with a side hustle teaching fitness classes, I thought certainly I am doing all of the right things and leading a healthy lifestyle, what could possibly be wrong?
That summer, I received a lab work order from my gynecologist, which was totally normal, but I had misplaced it shortly after and didnt think twice about it. Fast forward to my check-up in November with my doctor and he said your liver enzymes are very high. Is everything okay? We need to keep an eye on these.About three weeks later, I discovered my first huge blister on the top of my hand, which was incredibly painful. I went back into the doctor and they said, Have you touched anything that could have potentially given you an infection?Shortly after that, they started popping up all over the tops of my hands.
I was mortified by how my hands looked, not to mention, the pain I felt when anything came in contact with the tops of my hands. At this time, I was dating my now husband and we were preparing to leave the country to backpack in Spain. While on our trip, he proposed and I was ecstatic, but then the fear and embarrassment came over me. I knew what this meant I was going to have to answer to why my hands looked the way they did. Forget the ring, I was hands down mortified.
Upon returning to the states, I decided that I needed to take matters into my own hands. I started doing some research and decided to see a liver specialist. Thankfully, Dr. Ashfaq was first on my list and someone I am grateful for to this day. I began seeing him on a monthly basis while getting lab work done every two weeks. He knew my anxiety surrounding this and treated me with such compassion. Every time I would go in to see him, they would weigh me and I would feel even more beat down than before.
I was overcome with panic, worry, and anxiety because no one could pinpoint what was going on. He said, The typical candidate that has your liver enzyme levels is either (1) an alcoholic and/or (2) severely overweight.I was an anomaly. The more time went on and the more tests he ran, the more anxious I became.
While I still hadnt figured out what was causing the painful lesions on my hands, I determined that it would probably be in my best interest to see a dermatologist, who shall not be named. During my first encounter, they biopsied my hand and sent me on my way. I nursed an open wound for weeks and received no response from the doctor regarding the biopsy.
After some additional testing, Dr. Ashfaq ruled out Hepatitis C and determined that my porphyrin levels were off the charts, which was causing my elevated liver enzyme levels. Still not quite making the connection on how all of that would tie together, I continued to research on my own time. At this point, you could say research occupied a lot of my free time. Trying to connect these dots was very similar to how I feel trying to find a constellation in the sky.
It seemed that my life was a revolving door in and out of a doctors office. My hope, confidence, and resilience was slowly fading the more time went on. I feared not being in control when I was doing everything in my control to get better. I knew at this point I had to rely on the Lord and my support network to get me through these trying times. It wasnt going to be on Murphys time, it was going to be on His time.
You know, if you lose some weight, things in your body will start to change.
Stop eating carbs and stop drinking alcohol.
You need to lose 30 pounds and everything will improve.
Yeah, right.
After I had exhausted all avenues, Megan Lyons, a fellow fitness instructor and health/wellness coach, caught me outside of the gym as she was leaving. We shared one thing in common we were both fitness instructors but outside of that, I really didnt know Megan well. I am not sure what came over me in that moment, but I broke down to her. She showed me compassion and embraced me with words of strength and encouragement.
Thinking I had exhausted all avenues, she kindly offered to bring me in, take a look at my lab work, and see if there was any way she could help me. To this day, I still follow her supplement and nutrition regimen, which I have incorporated into my daily routine.
Though I had these tools in my tool kit, I was still trying to find someone to tell me what was going on with me.
As I continued to research the possibilities of what it could be, I thought it might be PCT based on what I had read online. Hundreds of Google searches later, I discovered the American Porphyria Foundation. After some clicking around the site and checking out the Board of Directors, I stumbled upon Dr. Karl Andersons name. Not only was he located in Texas, but he is one of the very few doctors in the country that specializes in PCT. I immediately picked up the phone, called his office, and left a message with someone hoping to hear back soon. Later that afternoon, I received a call back, from the man himself. In shock, I knew this was the Lord telling me something. This was His way of reigniting my fire to let me know that there would be answers, I just had to be patient and trust the process.
I connected Dr. Ashfaq and Dr. Anderson so they could discuss what was going on and together, they determined it was in my best interest to go see him in Galveston. Within two weeks, I was at UTMB meeting with Dr. Anderson and his staff.
It was confirmed that I had Porphyria Cutanea Tardae (PCT) caused by a mutation in the UROD gene. Because I was predisposed genetically for PCT, it couldve laid dormant my entire without ever rearing its ugly head. Though we dont know for certain what exactly caused it, I believe the birth control in large part was the major player.
After discussing all possible options, I determined that I would start my treatment with phlebotomies. I decided against the drug route initially because it was clear my liver wasnt functioning properly and I didnt want to send it into overdrive.
After two treatments, regular lab work, and maintaining a healthy lifestyle, I am now in remission. I now know what I need to do to manage my PCT moving forward. PCT doesnt define me, but it is a part of me. The scars on my hands are a constant reminder that I am greater than my circumstance.
I believe that the Lord bestows some of the biggest challenges to the strongest, most resilient people. While He might test you, it is important to always be steadfast in your faith because you never know what life might throw your way.
My diagnosis changed my perception about the world and made me realize how sweet life really is. I no longer take my health for granted. I am grateful, unlike many with PCT, to be able to wake up every day and live a relatively normal life. Now, at 28 years old, I am a bit more lighthearted and know that with the Lord, anything is possible. I am a better person because of my PCT.
My name is Allison Linner and I am seventeen. I came into this world on a cold, rainy day in early October, but my Dad said that the moment I was born, the sun peeked through the clouds, thus my nickname, Sunshine.
For some reason, my parents always knew there was something wrong with me. My life was dotted with strange symptoms, including lots of pain and lots of doctors for a girl my age. Each doctor diagnosed me with something else. When I hit puberty, things worsened. I had terrible episodes during which I would be doubled-over in front of the toilet, wanting to be unconscious because of the pain, but unwilling because I knew if I passed out I might choke on my own vomit. The toilet even became my pew. I fought to stay conscious while these other things were occurring. I could feel that my heartbeat was weak and irregular. Another strange symptom was that I was extraordinarily photosensitive. When ill, I would lie on my bed with my eyes shut, a wet washcloth over my eyes, the blinds closed tight, and I would still be irritated by light. This is one reason I thought I was just nuts. I even had times when I experienced paranoia and hallucinations. I would cry and pray hard. I wondered how my nickname could ever be Sunshine.
One of my most recent episodes was very hard. It was this past summer, and I decided to be the chaperone at an outdoor pool party my sister was attending. When I got out of the pool, one mom touched my face and said: Oh my Goshyoure so pale.I responded Yeah, I get that a lot. When we got back in the pool, I suddenly felt very ill and with Gods help I was able to get home and then get to the doctor.
I questioned him, In your professional opinion, do you think these symptoms that all of my different doctors have diagnosed over the years, are intertwined in some way?He said, No, there is no such disease that has all of your symptoms. But there had to be , I thought. I knew in my heart that each symptom was a puzzle piece and I realized that my family and I would have to play large roles in putting it together.
One day, my Dad and I talked a lot about what was happening. He promised me that I would find out what was wrong with me, and what was wrong with us, since we shared similar symptoms. At the end of our discussion, he jokingly said that maybe because we were so sensitive to light, we were vampires. He also said that there actually was a disease on which the legends were most likely founded. I thought I'd look it up, just for fun. Reading the list of symptoms was like reading the story of my life. It was actually a very spiritual experience, it felt like reading the scriptures. A few days later, I went to the doctor and asked him if he could order the tests. He said Sure, why not?While waiting on the obscure test results, I performed at home the '24-hour urine test.' Within a few hours, my urine was a deep purple-red hue. A few short days later, the tests came back positive for Hereditary Coproporphyria (HCP.)
This is a hard disease to have, there is no doubt about it. I know what pain is. But every day, I thank our Father in Heaven that I finally know what ails me; and that Im not crazy. Ive learned to cope with it daily. When I do have flare-ups that is probably when Im closer to God than any other time. It is so humbling. I am actually grateful for my disease because it does so much to compel me to be a better person.
When I turned 17 on October 10, my family gave me a card that has a cartoon sun smiling on it and it reads: Happy, Bright, and Lots of Fun.Inside, each member of my family wrote a little note to me. In my Dads message he wrote, You dont need the sun because you carry so much light inside you wherever you go.I guess Im still Sunshine after all.
NIH Captures Hereditary Coproporphyria (HCP)
Sunday - April 7, 2019 @ 07:00:01
Hereditary Coproporphyria (HCP)
HCP is an inherited autosomal dominant and a rare recessive disorder form of hepatic porphyria and less common than AIP, though latent HCP and HCP-carriers are being increasingly recognized. The prevalence of HCP in Denmark is approximately 2 per million. HCP is the least prevalent of the three principal types of acute porphyria: AIP, VP, and HCP. However, symptoms in HCP may be less frequent than in AIP or VP. Environmental or physiologic factors play a role in the pathogenesis of acute attack (Badminton and Elder, 2005).
2.2.1. Clinical features of HCP
HCP is an acute hepatic porphyria with neurovisceral symptoms, usually occurring as attacks that are indistinguishable from those in AIP (Bissell et al., 2012). Attacks typically start with low-grade abdominal pain that slowly increases over a period of hours or days, often with nausea and vomiting. Sometimes pain is predominantly in the back or extremities. A motor neuropathy may develop over a period of days or weeks if not treated. The neuropathy and weakness is often first noted proximally in the arms and legs, and may then progress distally. Respiratory insufficiency may result from impaired innervation of the diaphragm and respiratory muscles. Acute attacks are often associated with use of certain medications, caloric deprivation, and luteal phase increases in progesterone. About 20% of those with an acute attack report photosensitivity although bullae and skin fragility are much less common than in VP (Anderson et al., 2005 and 2014; Anderson and Lee, 2010). Of 46 individuals tested in Germany with acute HCP, 90% had abdominal pain; only 13% had cutaneous findings despite substantial overproduction of coproporphyrin (Kühnel et al 2000). An earlier British study of 111 individuals with HCP reported similar findings (Brodie et al., 1977).
Although disease-causing CPOX mutations occur equally in males and females, acute attacks are much more frequent in women, mainly between ages 16 and 45 years (the years of active ovulation). Chronic cutaneous HCP is suspected in individuals with bullae and fragility of light-exposed skin which result in depigmented scars; however, the cutaneous signs occur in only a minority of heterozygotes, even during an acute attack. In general, HCP is milder than AIP and is associated with fewer attacks. For example, in 32 members of an Australian family, 14 (including 10 adults) were determined to have HCP on the basis of a high fecal coproporphyrin III/I ratio and/or low lymphocyte CPOX enzyme activity, but only one had clinical symptoms of porphyria (Blake et al., 1992).
In rare homozygous cases of HCP, a CPOX mutation is inherited from each parent, with at least one of the mutant alleles expressing some enzyme activity. Characteristically, photosensitivity and chronic neurological manifestations begin in childhood, but acute attacks are seldom described (Schmitt et al., 2005).
2.2.2. Etiology and Pathogenesis of HCP
Like AIP and VP, HCP is inherited in an autosomal dominant manner with low penetrance. Heme production in most heterozygotes appears to be adequate. HCP results from coproporphyrinogen oxidase (CPOX) deficiency. At least 64 CPOX gene mutations involving all seven exons have been identified in different HCP families, including missense and nonsense mutations, large and small deletions and insertions, and splice site mutations (Rosipal et al., 1999; Schmitt et al., 2005; HGMD, 2014), as well as a few large deletions (Whatley et al., 2009; Barbaro et al., 2012). The enzyme functions as a homodimer, and some CPOX mutations may disrupt dimer formation (Lee et al., 2005). Homozygous cases of HCP have more severe CPOX deficiency, with onset of the disease in childhood.
CPOX catalyzes the 2-step decarboxylation of coproporphyrinogen III to protoporphyrinogen IX. Certain CPOX mutations release the substrate after one decarboxylation as harderoporphyrinogen, a tricarboxylate porphyrinogen that is then oxidized to the corresponding porphyrin. Homozygotes for these mutations have some distinct features, such as hemolysis early in life, whereas heterozygotes are similar to other HCP patients.
Because of reduced penetrance, many individuals with a CPOX mutation have no signs or symptoms of HCP. Given the rarity of acute attacks of HCP relative to AIP, it is suspected that only a small minority of CPOX heterozygotes express the clinical disease.
My name is Cassidy Clark and I have Erythropoietic Protoporphyria (EPP).
I am an 18-year-old high school senior. I like to think Im pretty funny, but Im mostly sarcastic - very sarcastic. I was diagnosed with EPP at age 15, but I have had symptoms since I was 6 years old.
Growing up, being outdoors was a huge part of my life. Me, my younger brother, who does not have EPP, and his best friend loved to play outside nonstop. Our favorite thing to do was to play front yard whiffle ball as well as swimming in our pool. We would stay outdoors as long as we could.
One random day when I was 6 years old, I came in and I started crying. My parents didn't know what was wrong. I would tell them over and over I was burning but just on the top of my baby toes. They didn't believe me, nor did they see anything wrong. They thought I was being a kidand complaining for no reason. However, the same burning sensation came back the next year and they knew then I wasnt lying about it. They knew something was wrong. We tried everything. We tried to put ice on it, put my toes in a cold bath, put a cold rag on it, but nothing would work. When I turned 10, it began to spread. It was still burning on my toes but also on my index finger and thumb. We didnt know what was wrong. When I was 12, it spread to the tops of my toes, my index finger, thumb, and then on the bridge of my nose to my cheekbones. I was in so much pain.
Throughout all my experiences, my parents would bring me to the hospitalhanding me over to the physician asking, Whats wrong with her?Unfortunately, they never had an explanation for it. The doctors would send us home. We felt hopeless, but we didnt stop there.
Things got worse when we went on a cruise to the Caribbean. I had just turned 14 years old. I remember we were on the island St. Maarten and we knew it had to do something with the sun, but we didnt think much of it. We ended up going to a beach and we thought getting an umbrella to put on the sand for when I'm at our spot was enough. We thought I would be also be fine in the water. And trust me we spent all day in the water. I remember when I got out of the water and we were drying off I was burning on my toes, that spot on my hands, that spot on my face and on top of my shoulders onto my back. It was the worst pain I have ever felt in my entire life. I remember on the way back to the boat I was balling my eyes out. As soon as I got onto the boat, I took a shower and it helped a little bit but not fully. One day we were looking through the TV channels and nothing good was on, so we clicked on a show called Dateline. This episode was about kids being allergicto the sun. You could hear crickets in the room as all of our jaws dropped. That night my mom looked it up to gather more information about it. We knew then, I had this rare disease. The next morning my mom called my doctor. My mom demanded to her that I had Porphyria. My doctor never heard of it before, but she did some research on it and she came to believe that I had this disease. She met with me but ended up sending me to a dermatologist. The dermatologist said in her 40 years of working about 20 people have come to her and said that they have this disease, but they never did, So, she sent me to Hasbro Children's Hospital. Unfortunately, my visit was not what Id hoped for. The staff didn't treat me like I was human, and the physician didn't believe I had this disease at all because of how rare it is. They made me wait hours and hours on end just because they thought I was wasting their time. It was awful. They ran a few blood tests and sure enough, everything was coming out positive. So, they transferred me to a specialist at Brigham and Women's Hospital in Boston. They ran a few tests and I was finally diagnosed on July 21st, 2016 at age 15.
We were so relieved! We finally knew what was wrong - but it doesnt end there. When I turned 16, I started burning everywhere and not only to the sun but also to lights as well. Then at 17, I became very sensitive to heat and I even get burned on cloudy days as well. Now finally, Im 18, and I always wear a hat and gloves. wear them inside as well. Its affected my life so much because Im a teenage girl. What do teenage girls do during the summer? Go to the beach and tan. I live 2 minutes away from the beach and I can never go. I want to be a normal girl, but I can't. Every time I do go in public, I have to wear a big hat that goes around, gloves, pants, and covered shoes. A lot of people laugh. And I mean a lot. People stare at me like I have some weird diseaseâ?¦ oh, wait I do. I don't really mind when little kids stare at me or ask their parents why I look so weird because they don't know any better. However, when adults stare and make rude comments. That hurts. They should know better. It's no different than them staring at someone if they're bald because they have cancer or someone in a wheelchair. I just really want to be normal. It mostly affects me at school. One of my favorite classes is gym. I love playing all the games because I'm very crazy and energetic. I love playing with my friends. But when they tell me the classes are going outside and I have to get a pass to go to the library, it breaks my heart. I want to play with all my friends, but I'm not allowed to. Also, if we have a fire drill or an evacuation, I have to be notified so I can "get ready" for it. That wastes their time to notify other kids, for example, ones that have hearing aids, so they don't get blasted. It doesn't only affect my education but other people's education as well because if I start having a reaction and I need the lights turned off, if they turn them off it may disrupt someone else. However, if they don't turn them off, I need to go to the nurse and sit in the nurse for the rest of the time, so I miss out on everything they're learning. The hardest thing about this disease is that nobody else in my family has it. None of my siblings, parents, grandparents, aunts, uncles, cousins. Nobody. I am the only one in my family. But I am trying to make the best out of this disease. I dont want to be known as a disease I want to be known as me. Since I do have this disease, I had the opportunity to go to Camp Sundown and it was the best week of my life. It was amazing finally meeting other kids who also live with this disease. They helped me understand that Im not alone in this. I have them. We will all get through this together.
Factual Overview of XLP from NIH
Saturday - April 6, 2019 @ 12:00:08
X-Linked Protoporphyria
Manisha Balwani, MD, MS, Joseph Bloomer, MD, Robert Desnick, MD, PhD, FACMG; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network.
X-linked protoporphyria (XLP) is characterized in affected males by cutaneous photosensitivity (usually beginning in infancy or childhood) that results in tingling, burning, pain, and itching within minutes of sun/light exposure and may be accompanied by swelling and redness. Vesicular lesions are uncommon. Pain, which may seem out of proportion to the visible skin lesions, may persist for hours or days after the initial phototoxic reaction. Photosensitivity usually remains for life. Multiple episodes of acute photosensitivity may lead to chronic changes of sun-exposed skin (lichenification, leathery pseudovesicles, grooving around the lips) and loss of lunulae of the nails. An unknown proportion of individuals with XLP develop liver disease. Except for those with advanced liver disease, life expectancy is not reduced. The phenotype in heterozygous females ranges from asymptomatic to as severe as affected males.
Diagnosis/testing.
Detection of markedly increased free erythrocyte protoporphyrin and zinc-chelated erythrocyte protoporphyrin is the most sensitive biochemical diagnostic test for XLP. Identification of pathogenic gain-of-functionvariants in ALAS2, the gene encoding erythroid specific 5-aminolevulinate synthase 2, confirms the diagnosis.
Management.
Treatment of manifestations: There is no FDA-approved treatment for this disease or specific treatment for the acute photosensitivity. The pain is not responsive to narcotic analgesics. Currently the only effective treatment is prevention of the painful attacks by avoidance of sun/light (including the long-wave ultraviolet light that passes through window glass) through use of protective clothing (e.g., long sleeves, gloves, wide-brimmed hats, protective tinted glass for cars and windows). Although topical sunscreens are typically not useful, some tanning products containing creams which cause increased pigmentation may be helpful. Oral Lumitene� (β-carotene) may improve tolerance to sunlight by causing mild skin discoloration due to carotenemia. Severe liver complications are difficult to treat: cholestyramine and other porphyrin absorbents (to interrupt the enterohepatic circulation of protoporphyrin and promote its fecal excretion) and plasmapheresis and intravenous hemin are sometimes beneficial. Liver transplantation can be a life-saving measure in individuals with severe protoporphyric liver disease; combined bone marrow and liver transplantation is indicated in those with liver failure to prevent future damage to the allografts.
Prevention of secondary complications: Vitamin D supplementation to prevent vitamin D insufficiency due to sun avoidance.
Surveillance: Monitoring of: hepatic function every 6-12 months and hepatic imaging if cholelithiasis is suspected; erythrocyte protoporphyrin levels (free and zinc-chelated), hematologic indices, and iron profile annually; vitamin D 25-OH levels.
Agents/circumstances to avoid: Sunlight and UV light; for those with hepatic dysfunction, drugs that may induce cholestasis (e.g., estrogens). For those with cholestatic liver failure, use of protective filters for artificial lights in the operating room to avoid phototoxic damage.
Evaluation of relatives at risk: If the ALAS2pathogenic variant has been identified in an affected family member, at-risk relatives can be tested as newborns or infants so that those with the pathogenic variant can benefit from early intervention (sun protection) and future monitoring for signs of liver dysfunction.
Therapies under investigation: Clinical trials are underway for afamelanotide, an α-melanocyte stimulating hormone analog, which increases skin pigmentation by increasing melanin production.
Genetic counseling.
XLP is inherited in an X-linked manner. Affected males transmit the pathogenic variant to all of their daughters and none of their sons. Women with an ALAS2 pathogenic variant have a 50% chance of transmitting the variant to each child. Carrier testing for at-risk relatives and prenatal testing for pregnancies at increased risk are possible if the pathogenic variant has been identified in an affected family member.
X-linked protoporphyria (XLP) should be suspected in individuals with the following findings:
Cutaneous photosensitivity, usually beginning in childhood
Burning, tingling, pain, and itching of the skin (the most common findings); may occur within minutes of sun/light exposure, followed later by erythema and swelling
Painful symptoms; may occur without obvious skin damage
Edema of the skin; may be diffuse and resemble angioneurotic edema
Absent or sparse vesicles and bullae (Note: The absence of skin damage [e.g., scarring], vesicles, and bullae often make it difficult to establish the diagnosis.)
Hepatic complications; may be life threatening, necessitating liver and/or bone marrow transplantation (BMT)
Testing
Detection of markedly increased free erythrocyte protoporphyrin and zinc-chelated erythrocyte protoporphyrin is the most sensitive biochemical diagnostic test for XLP (Table 1). Identification of a pathogenic gain-of-function variant in ALAS2, the gene encoding erythroid specific 5-aminolevulinate synthase 2, confirms the diagnosis (Table 2).
Table 1.
Biochemical Characteristics of X-Linked Protoporphyria (XLP)
Free protoporphyrin/zinc-chelated protoporphyrin: ratio 90:30 to 50:50 2, 3, 4
Protoporphyrins: not detectable
Protoporphyrin: normal or increased
Plasma porphyrins: increased 5
1.
Increased enzyme activity is due to ALAS2 pathogenic gain-of-function variants in exon 11. Note: Lymphocyte ferrochelatase activity is normal.
2.
Many assays for erythrocyte protoporphyrin or "free erythrocyte protoporphyrin" measure both zinc-chelated protoporphyrin and free protoporphyrin. Free protoporphyrin is distinguished from zinc-chelated protoporphyrin by ethanol extraction or HPLC.
3.
Protoporphyrins (usually zinc-chelated protoporphyrin) are also increased in lead poisoning, iron deficiency, anemia of chronic disease, and various hemolytic disorders, as well as in those porphyrias caused by biallelic pathogenic variants (e.g., harderoporphyria).
4.
In EPP, free protoporphyrin levels are elevated significantly as compared to zinc-chelated protoporphyrin (see Differential Diagnosis).
5.
Plasma total porphyrins are increased in porphyrias with cutaneous manifestations including XLP. If plasma porphyrins are increased, the fluorescence emission spectrum of plasma porphyrins at neutral pH can be characteristic and can distinguish XLP and EPP from other porphyrias. The emission maximum in XLP and EPP occurs at 634 nm.
Molecular Genetic Testing
Gene.ALAS2, which encodes the enzyme erythroid specific 5-aminolevulinate synthase 2, is the only gene in which pathogenic variants are known to cause XLP.
Table 2.
Molecular Genetic Testing Used in X-Linked Protoporphyria
Gene 1
Test Method
Pathogenic Variants Detected 2
Variant Detection Frequency by Test Method 3
Affected Males
Heterozygous Females
ALAS2
Sequence analysis
Sequence variants in exon 11 and in other coding and splicing regions 4
The ability of the test method used to detect a variant that is present in the indicated gene
4.
Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here.
5.
Eight out of eight families had one of two small deletions in exon 11. See Molecular Genetics.
To confirm/establish the diagnosis in a proband. For individuals with XLP-like photosensitivity, measurement of erythrocyte free and zinc-chelated protoporphyrin is the most sensitive and specific biochemical diagnostic test for XLP (Table 1). Note: It is essential to use an assay for erythrocyte protoporphyrin that distinguishes between free protoporphyrin and zinc-chelated protoporphyrin to differentiate XLP from EPP and several other conditions that may lead to elevation of erythrocyte protoporphyrins (see Table 1 footnotes 3 and 4).
However, sequencing of exon 11 of ALAS2 is the most accurate diagnostic method, especially for the detection of asymptomatic heterozygous females who may have normal erythrocyte protoporphyrin levels.
Carrier testing for at-risk relatives requires prior identification of the pathogenic variant in the family.
Note: (1) Carriers are heterozygotes for this X-linked disorder and may develop clinical findings related to the disorder. (2) Identification of female carriers requires either (a) prior identification of the pathogenic variant in the family or, (b) if an affected male is not available for testing, molecular genetic testing by sequence analysis.
Photosensitivity. Onset of photosensitivity is typically in infancy or childhood (with the first exposure to sun); in most individuals with XLP the photosensitivity remains for life.
Most individuals with XLP develop acute cutaneous photosensitivity within five to 20 minutes following exposure to sun or ultraviolet light. Photosensitivity symptoms are provoked mainly by visible blue-violet light in the Soret band, to a lesser degree in the long-wave UV region.
The initial symptoms reported are tingling, burning, and/or itching that may be accompanied by swelling and redness. Symptoms vary based on the intensity and duration of sun exposure; pain may be severe and refractory to narcotic analgesics, persisting for hours or days after the initial phototoxic reaction. Symptoms may seem out of proportion to the visible skin lesions. Vesicular lesions are uncommon.
Affected individuals are also sensitive to sunlight that passes through window glass, which does not block long-wave UVA or visible light.
Cutaneous manifestations. Multiple episodes of acute photosensitivity may lead to chronic changes of sun-exposed skin (lichenification, leathery pseudovesicles, grooving around the lips) and loss of lunulae of the nails. The dorsum of the hands is most notably affected.
Severe scarring is rare, as are hyper- or hypopigmentation, skin friability, and hirsutism.
Unlike in other cutaneous porphyrias, blistering and scarring rarely occur.
Hepatobiliary manifestations. Protoporphyrin is not excreted in the urine by the kidneys, but is taken up by the liver and excreted in the bile. Accumulated protoporphyrin in the bile can form stones, reduce bile flow, and damage the liver. Protoporphyric liver disease may cause back pain and severe abdominal pain (especially in the right upper quadrant).
The information on XLP and liver disease is limited. Based on one published report, it appears that the risk for liver dysfunction in XLP (observed in 5/31 affected individuals) is higher than the risk in EPP [Whatley et al 2008].
The information presented below is based on experience of liver disease in EPP [Bloomer 1988].
Gallstones composed in part of protoporphyrin may be symptomatic in individuals with XLP and need to be excluded as a cause of biliary obstruction in persons with hepatic decompensation. In most individuals underlying liver cirrhosis is already present; however, some may present with rapidly progressive cholestatic liver failure.
Life-threatening hepatic complications are preceded by increased levels of plasma and erythrocyte protoporphyrins, worsening hepatic function tests, increased photosensitivity, and increased deposition of protoporphyrins in hepatic cells and bile canaliculi. End-stage liver disease may be accompanied by motor neuropathy, similar to that seen in acute porphyrias. Comorbid conditions, such as viral hepatitis, alcohol abuse, and use of oral contraceptives, which may impair hepatic function or protoporphyrin metabolism, may contribute to hepatic disease in some [McGuire et al 2005].
Hematologic. Anemia and abnormal iron metabolism can occur in XLP. Mild anemia with microcytosis and hypochromia or occasionally reticulocytosis can be seen; however, hemolysis is absent or mild.
Bone marrow reticulocytes are thought to be the primary source of the accumulated protoporphyrin that is excreted in bile and feces. Most of the excess protoporphyrin in circulating erythrocytes is found in a small percentage of cells, and the rate of protoporphyrin leakage from these cells is proportional to their protoporphyrin content.
Erythrocyte protoporphyrin in XLP is both free and zinc-chelated in approximately equal proportions, although wide variation has been reported.
Ultraviolet light may cause free protoporphyrin to be released from the red cell even without disruption of the red cell membrane. In this manner, free protoporphyrin may then diffuse into the plasma, where it is bound to albumin and taken up by the endothelium of blood vessels.
The skin of persons with XLP is maximally sensitive to visible blue-violet light near 400 nm, which corresponds to the so-called Soret band (the narrow peak absorption maximum that is characteristic for protoporphyrin and other porphyrins). When porphyrins absorb light they enter an excited energy state. This energy is presumably released as fluorescence and by formation of singlet oxygen and other oxygen radicals that can produce tissue and vessel damage. This may involve lipid peroxidation, oxidation of amino acids, and cross-linking of proteins in cell membranes.
Photoactivation of the complement system and release of histamine, kinins, and chemotactic factors may mediate skin damage. Histologic changes occur predominantly in the upper dermis and include deposition of amorphous material containing immunoglobulin, complement components, glycoproteins, glycosaminoglycans, and lipids around blood vessels. Damage to capillary endothelial cells in the upper dermis has been demonstrated immediately after light exposure in this disease [Schneider-Yin et al 2000].
Individuals with autosomal recessive erythropoietic protoporphyria (EPP) seem predisposed to develop gallstones that are fluorescent and contain large quantities of protoporphyrin. While no information regarding gall stones in XLP is available, individuals with XLP are likely to be predisposed to cholelithiasis as well. This and other hepatobiliary complications relate to uptake and excretion of protoporphyrin by the liver [Bloomer 1988]. This dicarboxyl porphyrin is not soluble in aqueous solution and is therefore not excreted in urine.
Long-term observations of patients with protoporphyria generally show little change in protoporphyrin levels in erythrocytes, plasma, and feces. In contrast, severe hepatic complications, when they do occur, often follow increasing accumulation of protoporphyrin in erythrocytes, plasma, and liver. Iron deficiency and factors that impair liver function sometimes contribute. Enterohepatic circulation of protoporphyrin may favor its return and retention in the liver, especially when liver function is impaired. Liver damage probably results at least in part from protoporphyrin accumulation itself. As this porphyrin is insoluble, it tends to form crystalline structures in liver cells, can impair mitochondrial functions in liver cells, and can decrease hepatic bile formation and flow [Bloomer 1988, Anderson et al 2001].
Although sometimes considered a synonym for XLP, the term "erythropoietic protoporphyria, X-linked dominant" is incorrect and should not be used: in all X-linked metabolic disorders the phenotype in heterozygous females can range from asymptomatic to as severe as that seen in affected males.
Prevalence
The prevalence of XLP is unknown.
Based on studies from the UK, it appears that XLP accounts for about 2% of individuals with the EPP phenotype [Whatley et al 2010].
Preliminary studies from the US indicate that the percentage of individuals with XLP is higher in the US [Balwani et al 2013].
X-linked sideroblastic anemia, the only other phenotype known to be associated with pathogenic variants in ALAS2, is caused by pathogenic loss-of-function variants throughout ALAS2.
Erythropoietic protoporphyria, autosomal recessive(EPP) is caused by biallelic pathogenic variants in FECH (the gene encoding ferrochelatase). The photosensitivity and cutaneous manifestations are clinically indistinguishable from those seen in males with XLP. The only significant phenotypic difference is that only about 20%-30% of individuals with EPP have some degree of liver dysfunction, which is typically mild with slight elevations of the liver enzymes. Up to 5% may develop more advanced liver disease.
In EPP free protoporphyrin levels are elevated significantly as compared to zinc-chelated protoporphyrin (Table 3).
Table 3.
Biochemical Characteristics of Autosomal Recessive Erythropoietic Protoporphyria (EPP)
Deficient Enzyme
Enzyme Activity
Erythrocytes
Urine
Stool
Other
Ferrochelatase
<30% of normal
Protoporphyrin: >90% free, <10% zinc-chelated
Protoporphyrins: not increased
Protoporphyrin: normal or increased
Plasma porphyrins: increased
Possible additional genetic loci. It is presumed that additional loci may be responsible for the EPP phenotype (i.e., cutaneous photosensitivity and elevated erythrocyte protoporphyrins). Molecular epidemiology studies in the UK have identified biallelicFECH pathogenic variants or an ALAS2pathogenic variant in only 94% of unrelated individuals with the EPP phenotype [Whatley et al 2010].
To establish the extent of disease and needs of an individual diagnosed with X-linked protoporphyria (XLP), the following evaluations are recommended:
Assessment of erythrocyte protoporphyrin levels (free and zinc-chelated), hematologic indices, and iron profile if not performed as part of diagnostic testing
Assessment of hepatic function as well as imaging studies such as abdominal sonogram if cholelithiasis is suspected
Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
Acute photosensitivity. There is no FDA-approved treatment specific for this disease; furthermore, there is no specific treatment for the acute photosensitivity.
The pain is not responsive to narcotic analgesics.
Although several treatments have been proposed, most have been tried only in a single patient or a small number of patients. The only effective current treatment is prevention of the painful attacks by avoidance of sun/light.
Use of protective clothing including long sleeves, gloves, and wide brimmed hats is indicated.
Protective tinted glass for cars and windows prevents exposure to UV light. Gray or smoke-colored filters provide only partial protection.
Topical sunscreens are typically not useful; however, some tanning products containing creams which cause increased pigmentation may be helpful. Sun creams containing a physical reflecting agent (e.g., zinc oxide) are often effective but are not cosmetically acceptable to all.
Oral Lumitene� (β-carotene) (120180 mg/dL) may improve tolerance to sunlight if the dose is adjusted to maintain serum carotene levels in the range of 10-15 μmol/L (600800 μg/dL), causing mild skin discoloration due to carotenemia. The beneficial effects of β-carotene may involve quenching of singlet oxygen or free radicals. While oral beta-carotene has been used typically six to eight weeks before summer to reduce photosensitivity, its effectiveness may be limited [Minder et al 2009].
A systematic review of about 25 studies showed that the available data are unable to prove efficacy of treatments including beta-carotene, N-acetyl cysteine, and vitamin C [Minder et al 2009].
Hepatic disease. Treatment of hepatic complications, which may be accompanied by motor neuropathy, is difficult.
Cholestyramine and other porphyrin absorbents, such as activated charcoal, may interrupt the enterohepatic circulation of protoporphyrin and promote its fecal excretion, leading to some improvement [McCullough et al 1988]
Plasmapheresis and intravenous hemin are sometimes beneficial [Do et al 2002]
Liver transplantation has been performed as a life-saving measure in individuals with severe protoporphyric liver disease [McGuire et al 2005, Wahlin et al 2011b]. However, many transplant recipients experience a recurrence of the protoporphyric liver disease in the transplanted liver. Combined bone marrow and liver transplantation is indicated in patients with liver failure to prevent future damage to the allografts [Rand et al 2006]
Other. Iron supplementation may be attempted in persons with anemia and abnormal iron metabolism; close monitoring is warranted. Both clinical improvement and increased photosensitivity have been reported during iron replacement therapy in EPP [Holme et al 2007, Lyoumi et al 2007].
Whatley et al [2008] reported some evidence of diminished iron stores in males with XLP; in one patient with iron deficiency, iron repletion decreased protoporphyrin accumulation and corrected the anemia.
Prevention of Secondary Complications
Vitamin D supplementation is advised as patients are predisposed to vitamin D insufficiency due to sun avoidance.
Immunization for hepatitis A and B is recommended.
Surveillance
Annual assessment of erythrocyte protoporphyrin levels (free and zinc-chelated), hematologic indices, and iron profile is appropriate.
Hepatic function should be monitored every six to 12 months. Hepatic imaging studies such as an abdominal sonogram is indicated if cholelithiasis is suspected.
Vitamin D 25-OH levels should be monitored in all patients whether or not they are receiving supplements.
Agents/Circumstances to Avoid
The following are appropriate:
Avoidance of sunlight and UV light
In patients with hepatic dysfunction, avoidance of alcohol and drugs which may induce cholestasis (e.g., estrogens)
In patients with cholestatic liver failure, use of protective filters for artificial lights in the operating room to prevent phototoxic damage during procedures such as endoscopy and surgery [Wahlin et al 2008]
Evaluation of Relatives at Risk
If the ALAS2pathogenic variant has been identified in an affected family member, at-risk relatives can be tested as newborns or infants so that those with the pathogenic variant can benefit from early intervention (sun protection) and future monitoring for signs of liver dysfunction.
There is no information on pregnancy management in XLP. Based on experience with EPP pregnancy is unlikely to be complicated by XLP [Poh-Fitzpatrick 1997].
Therapies Under Investigation
Recent clinical trials with a subcutaneous insertion of a biodegradable, slow-released α-melanocyte stimulating hormone analog, afamelanotide, that increases pigmentation by increasing melanin appear promising for the treatment of EPP (see Differential Diagnosis) and XLP [Harms et al 2009, Minder et al 2009, Minder 2010].
In Europe Phase III trials have been completed and the drug is currently approved for the management of EPPin Italy and pending EMA approval for other countries in the European Union.
In the US, Phase II trials have been completed and Phase III trials are currently underway in order for the drug to receive FDA approval.
Genetic counseling is the process of providing individuals and families with information on the nature, inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members. This section is not meant to address all personal, cultural, or ethical issues that individuals may face or to substitute for consultation with a genetics professional. ED.
Mode of Inheritance
X-linked protoporphyria is inherited in an X-linked manner.
In a family with more than one affected individual, the mother of an affected male will have the pathogenic variant. Note: If a woman has more than one affected child and no other affected relatives and if the pathogenic variant cannot be detected in her leukocyte DNA, she has germline mosaicism. No data on the frequency of germline mosaicism in XLP are available.
If a male is the only affected family member (i.e., a simplex case), the mother may have the pathogenic variantor the affected male may have a de novo pathogenic variant. No data on the frequency of de novo pathogenic variants in XLP are available.
Parents of a female proband. A female with XLP may have a de novopathogenic variant or may have inherited an ALAS2 pathogenic variant from either parent; thus, if pedigree analysis reveals that the proband is the only affectedfamily member, it is reasonable to offer molecular genetic testing to both of her parents (starting with her mother) to determine risks to family members.
The risk to sibs depends on the genetic status of the parents.
If the father of the proband has an ALAS2pathogenic variant, he will transmit the variant to all of his daughters and none of his sons.
If the mother of the proband has an ALAS2pathogenic variant, the chance of transmitting it in each pregnancy is 50% to both males and females. Sons who inherit the pathogenic variant will be affected. Daughters may or may not be affected (see Penetrance).
If a male proband represents a simplex case (i.e., a single occurrence in a family) and if the pathogenic variantcannot be detected in the leukocyte DNA of the mother, the risk to sibs is low.
If a female proband represents a simplex case (i.e., a single occurrence in a family) and if the pathogenic variantcannot be detected in the leukocyte DNA of either parent, the risk to sibs is low.
Offspring of a male proband. Affected males transmit the pathogenic variant to all of their daughters and none of their sons.
Offspring of a female proband. Women with an ALAS2pathogenic variant have a 50% chance of transmitting the variant to each child.
Other family members of a proband. If a parent of the proband also has an ALAS2pathogenic variant, his or her family members may be at risk of having the pathogenic variant and of being affected, depending on their genetic relationship to the proband.
Note: Molecular genetic testing may be able to identify the family member in whom a de novopathogenic variantarose, information that could help determine genetic risk status of the extended family.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
The optimal time for determination of genetic risk and discussion of the availability of prenatal testing is before pregnancy.
It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk of having the ALAS2pathogenic variant.
DNA banking is the storage of DNA (typically extracted from white blood cells) for possible future use. Because it is likely that testing methodology and our understanding of genes, allelic variants, and diseases will improve in the future, consideration should be given to banking DNA of affected individuals.
Prenatal Testing and Preimplantation Genetic Diagnosis
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis. While decisions regarding prenatal testing are the choice of the parents, discussion of these issues is appropriate.
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. ED.
Data are compiled from the following standard references: gene from HGNC; chromosomelocus from OMIM; protein from UniProt. For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click here.
Gene structure. Alternatively spliced transcript variants of ALAS2 encode different isoforms (see Table A, Gene). The longest transcript has 11 exons.
Pathogenic variants
Eight families with XLP have been identified with one of two pathogenic variants (Table 4) [Whatley et al 2008]. Two small deletions, both in exon 11, introduce premature stop codons that predict truncation of the C-terminus of the enzyme.
Note on variant classification: Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
Note on nomenclature: GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product.ALAS2 encodes an erythroid-specific 5-aminolevulinate synthase; the normal isoform (NP_000023.2) has 587 amino acid residues, including a 49-amino acid transit peptide. The C-terminal amino acids encoded by exon 11 are believed to interact with the active site or other co-factors in a manner that regulates the activity of the enzyme.
Abnormal gene product. Truncation of the C-terminal amino acids or marked alteration in structure that confers an inability to interact normally results in increased ALAS2 enzyme activity [Whatley et al 2008]. This leads to the systemic accumulation of free and zinc-chelated protoporphyrins, particularly in erythroid and hepatic cells. The rate of 5-aminolevulinic acid formation is increased to such an extent that insertion of iron into protoporphyrin by FECH becomes rate limiting for heme synthesis, resulting in the accumulation of protoporphyrins [Whatley et al 2008].
Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of heme biosynthesis: X-linked sideroblastic anemias and the porphyrias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Vogelstein B, eds. The Metabolic and Molecular Bases of Inherited Disease. 8 ed. New York, NY: McGraw-Hill; 2001:2991-3062.
Balwani M, Doheny D, Bishop DF, Nazarenko I, Yasuda M, Dailey HA, Anderson KE, Bissell DM, Bloomer J, Bonkovsky HL, Phillips JD, Liu L, Desnick RJ. Loss-of-function ferrochelatase and gain-of-function erythroid 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and X-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria. Mol Med. 2013;19:2635. [PMC free article] [PubMed]
Bloomer JR. The liver in protoporphyria. Hepatology. 1988;8:4027. [PubMed]
Di Pierro E, Brancaleoni V, Tavazzi D, Cappellini M. C-terminal deletion in the ALAS2 gene and X-linked dominant protoporphyria. Haematologica. 2009;94 Suppl 2:315.
Do KD, Banner BF, Katz E, Szymanski IO, Bonkovsky HL. Benefits of chronic plasmapheresis and intravenous heme-albumin in erythropoietic protoporphyria after orthotopic liver transplantation. Transplantation. 2002;73:46972. [PubMed]
Harms JH, Lautenschlager S, Minder CE, Minder EI. Mitigating photosensitivity of erythropoietic protoporphyria patients by an agonistic analog of alpha-melanocyte stimulating hormone. Photochem Photobiol. 2009;85:14349. [PubMed]
Holme SA, Anstey AV, Badminton MN, Elder GH. Serum 25-hydroxyvitamin D in erythropoietic protoporphyria. Br J Dermatol. 2008;159:211. [PubMed]
Holme SA, Thomas CL, Whatley SD, Bentley DP, Anstey AV, Badminton MN. Symptomatic response of erythropoietic protoporphyria to iron supplementation. J Am Acad Dermatol. 2007;56:10702. [PubMed]
Lyoumi S, Abitbol M, Andrieu V, Henin D, Robert E, Schmitt C, Gouya L, de Verneuil H, Deybach JC, Montagutelli X, Beaumont C, Puy H. Increased plasma transferrin, altered body iron distribution, and microcytic hypochromic anemia in ferrochelatase-deficient mice. Blood. 2007;109:8118. [PubMed]
McCullough AJ, Barron D, Mullen KD, Petrelli M, Park MC, Mukhtar H, Bickers DR. Fecal protoporphyrin excretion in erythropoietic protoporphyria: effect of cholestyramine and bile acid feeding. Gastroenterology. 1988;94:17781. [PubMed]
McGuire BM, Bonkovsky HL, Carithers RL Jr, Chung RT, Goldstein LI, Lake JR, Lok AS, Potter CJ, Rand E, Voigt MD, Davis PR, Bloomer JR. Liver transplantation for erythropoietic protoporphyria liver disease. Liver Transpl. 2005;11:15906. [PubMed]
Minder EI. Afamelanotide, an agonistic analog of α-melanocyte-stimulating hormone, in dermal phototoxicity of erythropoietic protoporphyria. Expert Opin Investig Drugs. 2010;19:1591602. [PubMed]
Minder EI, Schneider-Yin X, Steurer J, Bachmann LM. A systematic review of treatment options for dermal photosensitivity in erythropoietic protoporphyria. Cell Mol Biol (Noisy-le-grand). 2009;55:8497. [PubMed]
Poh-Fitzpatrick MB. Human protoporphyria: reduced cutaneous photosensitivity and lower erythrocyte porphyrin levels during pregnancy. J Am Acad Dermatol. 1997;36:403. [PubMed]
Rand EB, Bunin N, Cochran W, Ruchelli E, Olthoff KM, Bloomer JR. Sequential liver and bone marrow transplantation for treatment of erythropoietic protoporphyria. Pediatrics. 2006;118:e18969. [PubMed]
Schneider-Yin X, Gouya L, Meier-Weinand A, Deybach JC, Minder EI. New insights into the pathogenesis of erythropoietic protoporphyria and their impact on patient care. Eur J Pediatr. 2000;159:71925. [PubMed]
Spelt JM, de Rooij FW, Wilson JH, Zandbergen AA. Vitamin D deficiency in patients with erythropoietic protoporphyria. J Inherit Metab Dis. 2010;33 Suppl 3:S14. [PubMed]
To-Figueras J, Ducamp S, Clayton J, Badenas C, Delaby C, Ged C, Lyoumi S, Gouya L, de Verneuil H, Beaumont C, Ferreira GC, Deybach JC, Herrero C, Puy H. ALAS2 acts as a modifier gene in patients with congenital erythropoietic porphyria. Blood. 2011;118:144351. [PubMed]
Wahlin S, Floderus Y, Stål P, Harper P. Erythropoietic protoporphyria in Sweden: demographic, clinical, biochemical and genetic characteristics. J Intern Med. 2011a;269:27888. [PubMed]
Wahlin S, Srikanthan N, Hamre B, Harper P, Brun A. Protection from phototoxic injury during surgery and endoscopy in erythropoietic protoporphyria. Liver Transpl. 2008;14:13406. [PubMed]
Wahlin S, Stal P, Adam R, Karam V, Porte R, Seehofer D, Gunson BK, Hillingsø J, Klempnauer JL, Schmidt J, Alexander G, O'Grady J, Clavien PA, Salizzoni M, Paul A, Rolles K, Ericzon BG, Harper P., European Liver and Intestine Transplant Association. Liver transplantation for erythropoietic protoporphyria in Europe. Liver Transpl. 2011b;17:10216. [PubMed]
Whatley SD, Ducamp S, Gouya L, Grandchamp B, Beaumont C, Badminton MN, Elder GH, Holme SA, Anstey AV, Parker M, Corrigall AV, Meissner PN, Hift RJ, Marsden JT, Ma Y, Mieli-Vergani G, Deybach JC, Puy H. C-terminal deletions in the alas2 gene lead to gain of function and cause x-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet. 2008;83:40814. [PMC free article] [PubMed]
Whatley SD, Mason NG, Holme SA, Anstey AV, Elder GH, Badminton MN. Molecular epidemiology of erythropoietic protoporphyria in the united kingdom. Br J Dermatol. 2010;162:6426. [PubMed]
The XLP contribution to GeneReviews was supported in part by the Porphyrias Consortium of the NIH-supported Rare Diseases Clinical Research Network (NIH grant: 5 U54 DK083909), including:
Dr Karl Anderson, University of Texas Medical Branch, Galveston, TX
Dr Montgomery Bissell, University of California, San Francisco, CA
Dr Herbert Bonkovsky, Carolinas Medical Center, Charlotte, NC
Dr John Phillips, University of Utah School of Medicine, Salt Lake City, UT
Erythropoietic protoporphyria (EPP) is a type of porphyria. Porphyrias are caused by an abnormality in the heme production process. Heme is essential in enabling our blood cells to carry oxygen and in breaking down chemical compounds in the liver. Erythropoietic protoporphyria is caused by pathogenic variants (mutations) in the FECHgene which lead to an impaired activity of ferrocheletase (FECH), an important enzyme in heme production. This results in the build-up of protoporphyrin in the bone marrow, red blood cells, blood plasma, skin, and eventually liver. Build up of protoporphyrin can cause extreme sensitivity to sunlight, liver damage, abdominal pain, gallstones, and enlargement of the spleen.[1] Inheritance is autosomal recessive.[1][2]
Treatment includes avoiding sun and UV light exposure, vitamin D supplements, creams for tanning, and using protective clothing. A medication known as Afamelanotide (Scenesse), a synthetic α-melanocyte stimulating hormone (a melanocyte is a skin cell that produces melanin, a skin-darkening pigment) analog was approved for treatment of EPP by the European Medicines Agency in 2014 and is awaiting approval in United States by the FDA. This medication increases pain-free sun exposure and has improved quality of life in those with EPP. Liver complications may be treated with cholestyramine and other porphyrin absorbents , plasmapheresis, a procedure where the liquid part of the blood, or plasma, is separated from the blood cells, and intravenous heme are sometimes beneficial. Liver transplantation may be required.[3][2]
Another type of porphyria, known as X-linked protoporphyria, is caused by a variation in the ALAS2 gene and have similar symptoms to erythropoietic protoporphyria when males are affected by EPP.[3]
This table lists symptoms that people with this disease may have. For most diseases, symptoms will vary from person to person. People with the same disease may not have all the symptoms listed. This information comes from a database called the Human Phenotype Ontology (HPO) . The HPO collects information on symptoms that have been described in medical resources. The HPO is updated regularly. Use the HPO ID to access more in-depth information about a symptom.
Erythropoietic protoporphyria is caused by mutations in the FECHgene.[3]
Last updated: 6/7/2016
In most cases, EPP is caused by mutations in the ferrochelatase (FECH) gene. Another type of protoporphyria caused by mutations in the delta-aminolevulinic acid synthase-2 (ALAS2) gene is known as X-linked protoporphyria (XLP). XLP have almost the same symptoms as the EPP in males, but appears to have a higher risk for liver problems than does EPP.[4][2][1]
EPP is inherited in an autosomal recessive manner. In most cases, affected individuals have one severe (loss-of-function) mutation that is inherited from one parent, and another weak (low-expression) mutation that is inherited from the other parent. In a small number of cases, an affected individual has two loss-of-function mutations.[4][3] When 2 carriers of an autosomal recessive condition have children, each child has a:
25% (1 in 4) chance to be affected
50% (1 in 2) chance to be an unaffected carrier like each parent
25% (1 in 4) chance to be unaffected and not be a carrier
Making a diagnosis for a genetic or rare disease can often be challenging. Healthcare professionals typically look at a persons medical history, symptoms, physical exam, and laboratory test results in order to make a diagnosis. The following resources provide information relating to diagnosis and testing for this condition. If you have questions about getting a diagnosis, you should contact a healthcare professional.
Testing Resources
The Genetic Testing Registry (GTR) provides information about the genetic tests for this condition. The intended audience for the GTR is health care providers and researchers. Patients and consumers with specific questions about a genetic test should contact a health care provider or a genetics professional.
The resources below provide information about treatment options for this condition. If you have questions about which treatment is right for you, talk to your healthcare professional.
Management Guidelines
Orphanet Emergency Guidelines is an article which is expert-authored and peer-reviewed that is intended to guide health care professionals in emergency situations involving this condition.
The American Porphyria Foundation offers a document that includes information about porphyria, types, testing, and treatment with Panhematin. Click the "document" link above to view these guidelines.
In the absence of liver failure, people with erythropoietic protoporphyria have normal life expectancies.[1] It is recommended to check the levels of the erythrocyte protoporphyrin (free and zinc-chelated), hematologic indices, and iron profile once a year. Liver function should also be monitored every six to 12 months. Vitamin D 25-OH levels should be monitored in all patients whether or not they are receiving supplements.[3]
If you need medical advice, you can look for doctors or other healthcare professionals who have experience with this disease. You may find these specialists through advocacy organizations, clinical trials, or articles published in medical journals. You may also want to contact a university or tertiary medical center in your area, because these centers tend to see more complex cases and have the latest technology and treatments.
If you cant find a specialist in your local area, try contacting national or international specialists. They may be able to refer you to someone they know through conferences or research efforts. Some specialists may be willing to consult with you or your local doctors over the phone or by email if you can't travel to them for care.
You can find more tips in our guide, How to Find a Disease Specialist. We also encourage you to explore the rest of this page to find resources that can help you find specialists.
Related diseases are conditions that have similar signs and symptoms. A health care provider may consider these conditions in the table below when making a diagnosis. Please note that the table may not include all the possible conditions related to this disease.
Conditions with similar signs and symptoms from Orphanet
Differential diagnosis includes phototoxic drug reactions, hydroa vacciniforme, solar urticaria, contact dermatitis, angioedema and, in some cases, other types of porphyria (see these terms).
Research helps us better understand diseases and can lead to advances in diagnosis and treatment. This section provides resources to help you learn about medical research and ways to get involved.
Clinical Research Resources
ClinicalTrials.gov lists trials that are related to Erythropoietic protoporphyria. Click on the link to go to ClinicalTrials.gov to read descriptions of these studies.
Please note: Studies listed on the ClinicalTrials.gov website are listed for informational purposes only; being listed does not reflect an endorsement by GARD or the NIH. We strongly recommend that you talk with a trusted healthcare provider before choosing to participate in any clinical study.
Patient Registry
The Porphyrias Consortium is a team of doctors, nurses, research coordinators, and research labs throughout the U.S., working together to improve the lives of people with this condition through research. The Porphyrias Consortium has a registry for patients who wish to be contacted about clinical research opportunities.
Support and advocacy groups can help you connect with other patients and families, and they can provide valuable services. Many develop patient-centered information and are the driving force behind research for better treatments and possible cures. They can direct you to research, resources, and services. Many organizations also have experts who serve as medical advisors or provide lists of doctors/clinics. Visit the groups website or contact them to learn about the services they offer. Inclusion on this list is not an endorsement by GARD.
RareConnect has an online community for patients and families with this condition so they can connect with others and share their experiences living with a rare disease. The project is a joint collaboration between EURORDIS (European Rare Disease Organisation) and NORD (National Organization for Rare Disorders).
Living with a genetic or rare disease can impact the daily lives of patients and families. These resources can help families navigate various aspects of living with a rare disease.
Financial Resources
The HealthWell Foundation provides financial assistance for underinsured patients living with chronic and life-altering conditions. They offer help with drug copayments, deductibles, and health insurance premiums for patients with specific diseases. The disease fund status can change over time, so you may need to check back if funds are not currently available.
These resources provide more information about this condition or associated symptoms. The in-depth resources contain medical and scientific language that may be hard to understand. You may want to review these resources with a medical professional.
Where to Start
DermNet NZ is an online resource about skin diseases developed by the New Zealand Dermatological Society Incorporated. DermNet NZ provides information about this condition.
Genetics Home Reference (GHR) contains information on Erythropoietic protoporphyria. This website is maintained by the National Library of Medicine.
MedlinePlus was designed by the National Library of Medicine to help you research your health questions, and it provides more information about this topic.
The National Human Genome Research Institute's (NHGRI) website has an information page on this topic. NHGRI is part of the National Institutes of Health and supports research on the structure and function of the human genome and its role in health and disease.
In-Depth Information
Medscape Reference provides information on this topic. You may need to register to view the medical textbook, but registration is free.
The Monarch Initiative brings together data about this condition from humans and other species to help physicians and biomedical researchers. Monarchs tools are designed to make it easier to compare the signs and symptoms (phenotypes) of different diseases and discover common features. This initiative is a collaboration between several academic institutions across the world and is funded by the National Institutes of Health. Visit the website to explore the biology of this condition.
Online Mendelian Inheritance in Man (OMIM) is a catalog of human genes and genetic disorders. Each entry has a summary of related medical articles. It is meant for health care professionals and researchers. OMIM is maintained by Johns Hopkins University School of Medicine.
Orphanet is a European reference portal for information on rare diseases and orphan drugs. Access to this database is free of charge.
PubMed is a searchable database of medical literature and lists journal articles that discuss Erythropoietic protoporphyria. Click on the link to view a sample search on this topic.
#PAW2019 Starts April 6th- 13th
Wednesday - March 27, 2019 @ 09:47:28
Welcome to #PAW2019 Runs April 6th-13th Watch every day for members stories, Factual Information from Experts Learn about all of the Porphyrias! Games & Prizes on the Open Main APF FB Page! Answer correctly and you can win some APF swag Check out all the NEW APF Merchandise items on the APF store www.porphyriafoundation.org
APF Merchandise
Friday - March 15, 2019 @ 06:30:02
Please get your APF Merchandise today all sizes have been updated in the line item description. Prices are now on clearance. If you see a zero in the size you want we will NOT be ORDERING and ask that you choose another item. Watch out in the next week for the new tshirts-umbrellas-super nice-APF bags.
https://porphyriafoundation.org/store
Thank you for supporting the APF
& Porphyria Warrior!
USA CANADA EUROPE UK ASIA PAKISTAN RUSSIA We shipp anywhere for an additional fee once an order is placed we will contact you via FB or Email to get your permission of international fee
My story is probably not much different than most. I was misdiagnosed most of my life, sent to psychologists because physicians just couldn't find a cause for my illness. Finally, after an incorrect Lupus diagnosis and a week of testing, I received my Variegate Porphyria diagnosis.
As a child growing up in Tampa, FL, I would constantly complain that the sun hurt my skin and my eyes but "children" were not supposed to wear sun glasses. Today I am rarely seen without them, day or night. I craved sweets and of course "children" should not eat too much sugar! I went to a boarding school most of my life and "they" had the same rules about sun protection and sweets the same as my parents. For reasons like this, I have always wanted to write a book "If you Can't See itIt Must Not Be Real" for all of "us" who are ill but it can't be seen, and so it must be in our "heads"!!
How might I be different? I have always known I am a survivor in life and have tried to maintain a positive attitude. At a very early age, I chose the "high road". I have pushed myself both mentally and physically to go beyond and not make my disease a focus in my life. I worked hard to receive a good education, eventually owned a successful cooperation, raised three step children and married twice. Now I ride and show horses (jumpers), work out daily and volunteer whenever I can (I have recently been helping women coming out of prison to assimilate back into the business world) and continue to look for areas and people to add to my life.
I have not had the best experiences with physicians. I was made worse by a physician claiming I would NEVER meet anyone else with my disease. He was like many physicians, who know very little about the disease and read only one chapter in medical school. Nonetheless, he was a self-proclaimed expert on porphyria with his own "treatments." I am alive today after surviving his dilation toxicity for three years and having a gland removed by his surgeon friend. This left me with permanent double vision at three and half feet out. Thus, reading is almost impossible for me other than my large computer screen.
Next, I broke most of my fingers and had another serious problem. I am going through implant surgery now because of mishandling by a dentist. Then I was left bleeding to death in "after surgery" care, overdosed with morphine, followed by a surgeon who was drunk and performed ovarian surgery incorrectly. I could go on, but I suffice to write this to let you know "we" must be our own advocates in health care. I still seek the good in all I meet and do believe there are wonderfully talented and gifted physicians, but my experiences have certainly caused me to be vigilant over my own care. As for porphyria, I thank God for Dr. Anderson, his colleagues and Desiree.
My attitude is to count my blessings, allow myself some few "moments" of self pitypick myself up and move forward. We all have challenges in life and sometimes hurt so badly that it is hard to live another day--but we do. We need to share information and stories, support to each other and always keep our sense of humor. I try to look at what I have rather than what I don't have.
In the end it will always be our "attitudes" that drive our lives. As a very wise man long ago told me, "You have two choices in the morning when you start your day" for whatever reason, hearing him tell me on that day changed my life, and I say this to all of you hoping I can make the same difference. Good luck and God Bless!
Porphyria Post
Wednesday - March 13, 2019 @ 06:00:03
Porphyria Post
FDA Announces Interim Commissioner
On Tuesday, the Food and Drug Administration announced that Dr. Ned Sharpless will step in as acting Commissioner following the resignation of Dr. Scott Gottlieb last week.
Sharpless is an accomplished researcher, oncologist and administrator. He was named director of the National Cancer Institute in October of 2017 following his position as director of the University of North Carolina Lineberger Comprehensive Cancer Center.
Alex M. Azar III, secretary of health and human services, stated that this position is temporary and the FDA is continuing their search for a permanent commissioner.
FIND YOUR SHADOW 2019 applications are due in just 2 days!!
Don't miss this opportunity for a trip to Disney for your family!!
Click here to apply TODAY!
URGENT! URGENT! PAIN COMMENTS NEEDED ASAP!!!
Do you experience pain due to Acute Porphyria? Please take 5 minutes to submit a comment TODAY to help the federal government develop official guidelines. A government task force is addressing how to manage pain in the face of the opioid crisis. Their recommendations will become the guidelines for all government agencies. The American Porphyria Foundation has been very involved in this committee. Now it is your turn to make public comment. It doesnt need to be long or perfect. Time is of the essence.
Your comment should include: - Your diagnosis Sample: I was diagnosed with Acute Intermittent Porphyria fifteen years ago at the age of twenty-six.
- Description of your pain Sample: worst pain in humankind,torture,feels like a thousand flaming swords,inhumane,feels like coals burning in my abdomen,etc.
- Unsafe drugs trigger an Acute Porphyria attack Sample: Acute Intermittent Porphyria is a pharmacogenetic disease. If I take an unsafe drug, it will cause an attack which includes extreme pain.
- That opioids are the ONLY safe drug that helps to manage the severe pain of acute porphyria. Sample: My physician has prescribed and managed safe opioid use for my disease for 10 years.
- Any personal details you would like to share Sample: I have been called a drug-seeker for requesting opioid pain management even with a diagnosis. I have been sent away from emergency rooms and hospitals.
Porphyria Awareness Week 2019 is less than a month away and we want to hear what YOU are doing in your community to spread the word!
Jason Sumney (AIP) is raising awareness for Porphyria and the APF at the Pittsburgh Vintage Mixer on April 6th, 2019. Megan Dunn- Tonhaeuser and her children are purchasing wristbands and handing our Porphyria Fact Sheets at their school.
The opportunities are limitless so email porphyrus@porphyriafoundation.com and let us know what you have planned for PAW 2019!
Acute Porphyria membersYour help is needed!
Attention All Acute Patients!!
Have you had trouble getting PANHEMATIN in a hospital for ANY REASON - please contact the APF on 866-APF-3635 or email porphyrus@porphyriafoundation.org.
We need to hear your story. The APF is gathering your patient stories to prove that this is an issue across the US and to advocate for access to treatment on a federal level.
Volunteers Needed!
The APF will be exhibiting at the 2019 ACMG Annual Meeting in Seattle, WA April 3-5 AND Digestive Disease Week in San Diego, CA May 19-21. Are you local to the area and interested in volunteering at our booth? If so, please contact Edrin Williams, Director of Patient Services, for additional information at edrinw@porphyriafoundation.org or 301.347.7166.
This is an incredible opportunity to raise awareness about porphyria to those with little knowledge about this rare disease.
RSVP Today
Patient Education and Support Meeting- Ann Arbor
LOCATION Holiday Inn DATE AND TIME 03/16/19 1:00pm - 03/16/19 3:00pm 3600 Plymouth Road Ann Arbor, MI Room: Wolverine A Family and friends are welcome to attend!
I'll be there!
Maybe
I can't make it Patient Education and Support Meeting- Seattle
LOCATION Seattle Public Library- Central DATE AND TIME 04/04/19 5:30pm - 04/04/19 7:30pm 1000 Fourth Avenue Seattle, WA Room: Washington Mutual Level: 4 Family and friends are welcome to attend!
I'll be there!
Maybe
I can't make it
Patient Education and Support Meeting- Boston
LOCATION Morrill Memorial Library DATE AND TIME 04/06/19 2:00pm - 04/06/19 4:00pm 33 Walpole Street Norwood, MA Room: Simoni Family and friends are welcome to attend!
I'll be there!
Maybe
I can't make it
Get Involved
â?¢ Research â?¢
â?¦ Donate â?¦
â?¢ Visit our Website â?¢
Contact Information
Is your contact information up to date? If not please give us a call @ 866-APF-3635 or Email to porphyrus@porphyriafoundation.com
If you need to consult with a Porphyria specialist and are unable to do so because of the distance to a Porphyria center, you now have the opportunity to consult electronically.
A Porphyria specialist and member of the APF Scientific Advisory Board, Dr. Karl Anderson, will be consulting with Porphyria patients via a telemedicine service from the General Clinical Research Center of the University of Texas Medical Branch at Galveston.
To access this service, you need to first locate a video teleconferencing facility in your community. These facilities are often found in libraries, businesses, universities and research centers. Unfortunately, neither the APF nor Dr. Anderson can provide you with a list of facilities with teleconferencing capabilities. Next, call Dr. Karl Anderson (409) 772-4661 to arrange for an appointment and to find out what information you might need to have ready prior to the telemedicine visit.
During the teleconference, Dr. Anderson will discuss your case with you just as he would during a regular office visit. This project was funded in part by a grant from the American Porphyria Foundation.
Physician Information Kit Available
Upon request, the APF will send a comprehensive physician education packet on the acute porphyrias to physicians. It includes educational materials on the diagnosis and management of the acute porphyrias; information on biochemical and genetic testing; Panhematin treatment for the acute porphyrias; and a recent article from the Annals of Internal Medicine on diagnosis and management of the acute porphyrias. Please call our office at (301) 347-7166 to order the kit.
In addition, the APF will also send information on PCT and EPP to physicians upon request. Please call our office at (301) 347-7166 to order the packet.
These packets are delivered via USPS, for physicians only.
Porphyria Slide Set
This PowerPoint slide set provides valuable information about porphyria, including discussion covering porphyria types, causes, diagnosis, and treatment protocols. It also includes vital information focusing on Panhematin (hemin for injection).
Last summer I was babysitting my four grandchildren at my daughter's house while they were on vacation, I noticed a large blister on my hand and thought I had splattered grease on it. After I went home a week later, I had more blisters on my hands of different sizes. I had a really big one on my right hand, and every time I shook hands it would hurt.
A friend suggested that I should sterilize a needle and drain it. I did so and put Neosporin on it as well. Needless to say it got infected, so I went to the Dr. and he started running tests on me. After several tests, he found my liver enzymes were high, so he sent me to a dermatologist who did a biopsy of a small blister and had me do the 24-hour urine test. That is when I found out I had Porphyria Cutanea Tarda (PCT).
Next I started the phlebotomies. On Dec. 1st, the 8th time the Dr. did the blood draw, I got what I thought was the flu and was so sick, I could not go anywhere without falling back on the couch, out of breath and I was very weak. My best friend told me to call the Dr. and she would take me as I couldn't drive. I did call and she took me to see my regular doctor. His nurse walked in and took one look at me, and said I was anemic. The doctor checked me and said I needed iron. I told him I couldn't because of my porphyria.
Later my other Dr. called me after they all talked and said that I would just have to rest. I eventually started felling better.
In January of this year, the doctor took another blood test that was better. Now I am going in every three months for my blood checks. The scars on my hands are fading slowly. My granddaughter says your "owies" are better and I can hold your hands, and my skin is not so fragile as it was, although I am a grandma and most grandmas have fragile skin, it should not be that fragile yet.
I was noticing some of the other symptoms of PCT, and I just went to the Dr. complaining of pain in my arm, hands, and insomnia.
Where I live they don't know much about porphyria. My life is so much better now, but I still stay out of the sun, avoid alcohol and estrogen, and yes my problems could be much worse. When I think about the plight of other people, I thank God every night for my health. Good luck to everyone.
AHP- URGENT! URGENT! PAIN COMMENTS NEEDED ASAP!
Wednesday - March 6, 2019 @ 06:30:05
URGENT! URGENT! PAIN COMMENTS NEEDED ASAP!
Make a public comment today through this link: https://www.regulations.gov/comment?D=HHS-OS-2018-0027-0001 These comments will help inform the federal government about the needs of patients with Acute Porphyria as they develop federal guidelines. Patients, caregivers, and physicians can all comment. Share this link with others.
- Why you are writing
- Your diagnosis
- Description of your pain
- Unsafe drugs trigger an Acute Porphyria attack
- That opioids are the ONLY safe drug that helps to manage the severe pain of acute porphyria.
- Any personal details you would like to share
Don't wait - take 5 minutes and do this right away!
Over the years this time honored tradition has inspired porphyria patients to go out and educate their community on porphyria. Over the years many great ideas have been put into action such as, in 2014 when Amanda Rich made purple bracelets for porphyria survivors or in 2015 when Victor Mejias hosted a fishing tournament for the APF. In 2018, Cassie Tucker and her son created a T-shirt campaign to raise awareness for EPP and donated part of the proceeds to the APF! This is the time to use your hobbies and talents to spread the word about porphyria. There is no limit to what you can accomplish!
What does 2019 hold? Murphy McNutt (PCT) is organizing a fundraiser for the APF at her fitness studio. She is planning a "ride" with the support of the studio owner and plans to have riders wear purple! Check out Murphy's story on the APF website Member Stories, visit https://porphyriafoundation.org/member-stories/murphy-mcnutt.
Over the next few weeks, the APF will be sharing resources and tools for you to create awareness in your community. Need ideas? Give the APF office a call on 866-APF-3635.
Let us know what YOU are planning and together let's PAINT THE WORLD PURPLE!
PSA
Friday - February 15, 2019 @ 16:49:42
Like shards of glass under the skin!
It feels like millions of broken pieces of glass throughout My body.
To move is just sheer pain.
If you feel like I do, remain optimistic, if you dont have answers to your health questions, keep searching, stay patient yet persistent!
As my Nurse told me yesterday, speak up speak loud and insist on the best professional care.
Hours turn into days, days turn in weeks, months and years dont waste your life away.
Dont give up! That is my resolve in my life what is yours?
Porphyria Post
Thursday - February 14, 2019 @ 13:09:27
February 16, 2019
Coming to a city near you!!
Patient Education and Support Meetings
Throughout 2019 the APF will be hosting a dozen Patient Education and Support Meetings across the country. This is a remarkable opportunity to meet fellow porphyria patients and families to share similar experiences. Porphyria experts will also be present at each event either in person or via Skype.
RSVP below to our Phoenix, Ann Arbor, Seattle, Boston, Salt Lake City, and Placentia, CA meetings.
Be on the lookout for more details regarding the upcoming meetings below!
Birmingham, AL
New York, NY
Winston- Salem, NC
Chicago, IL
Orlando, FL
Interested in hosting a meeting in your town? Contact Edrin Williams, Director or Patient Services, at edrinw@porphyriafoundation.org.
RSVP Today
Patient Education and Support Meeting- Phoenix
LOCATION
Holiday Inn & Suites Phoenix Airport
DATE AND TIME
02/23/19 1:00pm - 02/23/19 3:00pm
3220 S. 48th Street Phoenix, AZ Room: Scottsdale Family and friends are welcome to attend!
Patient Education and Support Meeting- Ann Arbor
LOCATION
Holiday Inn
DATE AND TIME
03/16/19 1:00pm - 03/16/19 3:00pm
3600 Plymouth Road Ann Arbor, MI Room: Wolverine A Family and friends are welcome to attend!
Patient Education and Support Meeting- Seattle
LOCATION
Seattle Public Library- Central
DATE AND TIME
04/04/19 5:30pm - 04/04/19 7:30pm
1000 Fourth Avenue Seattle, WA Room: Washington Mutual Level: 4 Family and friends are welcome to attend!
Patient Education and Support Meeting- Boston
LOCATION
Morrill Memorial Library
DATE AND TIME
04/06/19 2:00pm - 04/06/19 6:00pm
33 Walpole Street Norwood, MA Room: Simoni Family and friends are welcome to attend!
Patient Education and Support Meeting- Placentia, CA
LOCATION
Home of Louise Schlosser Braun
DATE AND TIME
04/27/19 11:00am - 04/27/19 1:00pm
Refreshments will be provided RSVP for address and additional Information Family and friends are welcome to attend!
Patient Education and Support Meeting- Salt Lake City
LOCATION
University of Utah- University Guest House and Conference Center
DATE AND TIME
05/31/19 5:00pm - 05/31/19 7:00pm
110 S. Fort Douglas Blvd Salt Lake City, UT Room: Bonneville Family and friends are welcome to attend!
Contact Information
Is your contact information up to date?
If not please give us a call @ 866-APF-3635 or Email to porphyrus@porphyriafoundation.com
American Porphyria Foundation 1.866.APF.3635 porphyriafoundation.org
STAY CONNECTED
"Remember.Research is the Key to Your Cure!"
NEW- Porphyria POST
Thursday - February 14, 2019 @ 11:34:18
Porphyria Post
OFFICE CLOSURE
The APF office will be closed on Monday, February 18, 2019 in observation of President's Day. Normal hours of 9:00am-5:00pm EST will resume Tuesday, February 19, 2019.
Porphyria Awareness Week - April 6-13, 2019
Over the years this time honored tradition has inspired porphyria patients to go out and educate their community on porphyria. Over the years many great ideas have been put into action such as, in 2014 when Amanda Rich made purple bracelets for porphyria survivors or in 2015 when Victor Mejias hosted a fishing tournament for the APF. In 2018, Cassie Tucker and her son created a T-shirt campaign to raise awareness for EPP and donated part of the proceeds to the APF! This is the time to use your hobbies and talents to spread the word about porphyria. There is no limit to what you can accomplish!
What does 2019 hold? Murphy McNutt (PCT) is organizing a fundraiser for the APF at her fitness studio. She is planning a "ride" with the support of the studio owner and plans to have riders wear purple! Check out Murphy's story on the APF website Member Stories, visit https://porphyriafoundation.org/member-stories/murphy-mcnutt.
Over the next few weeks, the APF will be sharing resources and tools for you to create awareness in your community. Need ideas? Give the APF office a call on 866-APF-3635.
Let us know what YOU are planning and together let's PAINT THE WORLD PURPLE!
Acute Porphyria membersYour help is needed!
Attention All Acute Patients!!
Have you had trouble getting PANHEMATIN in a hospital for ANY REASON - please contact the APF on 866-APF-3635 or email porphyrus@porphyriafoundation.org.
We need to hear your story. The APF is gathering your patient stories to prove that this is an issue across the US and to advocate for access to treatment on a federal level.
Acute Porphyria Survey
A research company has initiated an online survey to get a deeper understanding of patientsexperiences living with acute porphyria. The study can be taken on a laptop or desktop computer and will take approximately 30 minutes to complete. Participants will be screened to ensure they meet the criteria listed below.
Participants will receive a $50 stipend!
Criteria:
1. Are 18 years of age or older
2. Have been diagnosed with Acute Hepatic Porphyria (specific types include Acute Intermittent Porphyria, Variegate Porphyria, Hereditary Coproporphyria).
3. Own and are comfortable using a laptop or a desktop computer
4. Have internet access
Please call the APF at 866-APF-3635 to participate in the survey.
Volunteers Needed!
The APF will be exhibiting at the 2019 ACMG Annual Meeting in Seattle, WA April 3-5 AND Digestive Disease Week in San Diego, CA May 19-21. Are you interested in volunteering at our booth? If so, please contact Edrin Williams, Director of Patient Services, for additional information at edrinw@porphyriafoundation.org or 301.347.7166.
This is an incredible opportunity to raise awareness about porphyria to those with little knowledge about this rare disease.
RSVP Today
Patient Education and Support Meeting- Phoenix
LOCATION
Holiday Inn & Suites Phoenix Airport
DATE AND TIME
02/23/19 1:00pm - 02/23/19 3:00pm
3220 S. 48th Street Phoenix, AZ Room: Scottsdale Family and friends are welcome to attend!
Patient Education and Support Meeting- Ann Arbor
LOCATION
Holiday Inn
DATE AND TIME
03/16/19 1:00pm - 03/16/19 3:00pm
3600 Plymouth Road Ann Arbor, MI Room: Wolverine A Family and friends are welcome to attend!
Patient Education and Support Meeting- Seattle
LOCATION
Seattle Public Library- Central
DATE AND TIME
04/04/19 5:30pm - 04/04/19 7:30pm
1000 Fourth Avenue Seattle, WA Room: Washington Mutual Level: 4 Family and friends are welcome to attend!
Contact Information
Is your contact information up to date?
If not please give us a call @ 866-APF-3635 or Email to porphyrus@porphyriafoundation.com
American Porphyria Foundation 1.866.APF.3635 porphyriafoundation.org
STAY CONNECTED
"Remember.Research is the Key to Your Cure!"
PSA
Thursday - February 7, 2019 @ 12:47:42
Someone who is physically dependent on opioids as a result of the treatment of pain but who is not craving the drugs is not addicted.
Dr. Scott Gottlieb, FDA Commissioner
NEW~ Porphyria Post
Friday - February 1, 2019 @ 16:23:50
Porphyria Post
Porphyria Awareness Week - April 6-13, 2019
Over the next few weeks, the APF will be sharing resources and tools for you to create awareness in your community. Need ideas? Give the APF office a call on 866-APF-3635.
In 2018, countless members raised awareness through conversations, activities, fundraisers and more!! Leann Cook held a raffle at a local store for a donated Yeti cooler with signage about porphyria. Griffins sixth grade class all wore purple to honor and remember his mom. Louise Schlosser Braun hosted a table full of APF brochures at a local hospital to train Emergency Room staff and interns. Allison Stuhlsatz wrote for her local blog, the Wichita Moms Blog. Justin and Holly Hamilton were featured in their local newspaper, the Wauneta Breeze, about living with CEP. Dr. Bruce Wang (Porphyria Expert, UCSF) and Mary Schloetter did 17 back-to-back satellite media interviews all which were written or shared in newspapers and local media across the country. Jared Ulmer, EPP, videoed a visit to a middle school science class where he taught them about his genetic condition. Jenna Steel, age 17, created a video about living with EPP. Morgan McKillop, age 8, was featured in a short film about living with a photosensitive condition. Jennifer Becks sister held a fundraising event at her workplace. Dr. Amy Dickey, EPP, held a birthday/awareness fundraising event on Facebook. Sharon Dill (VP) created a short video about living with porphyriait was distributed to family and friends to raise awareness about the everyday burden. We even had a student at Eastern Florida State College, Charissa Strandberg, complete a genetics project on porphyria presented to class (wearing purple!) along with brochures.
What does 2019 hold? Murphy McNutt (PCT) is organizing a fundraiser for the APF at her fitness studio. She is planning a "ride" with the support of the studio owner. She plans to have riders wear purple! Check out Murphy's story on the APF website Member Stories, visit https://porphyriafoundation.org/member-stories/murphy-mcnutt
Let us know what YOU are planning!
Acute Porphyria membersYour help is needed!
Attention All Acute Patients!!
Have you had trouble getting PANHEMATIN in a hospital for ANY REASON - please contact the APF on 866-APF-3635 or email porphyrus@porphyriafoundation.org.
We need to hear your story. The APF is gathering your patient stories to prove that this is an issue across the US and to advocate for access to treatment on a federal level.
Acute Porphyria Survey
A research company has initiated an online survey to get a deeper understanding of patientsexperiences living with acute porphyria. The study can be taken on a laptop or desktop computer and will take approximately 30 minutes to complete. Participants will be screened to ensure they meet the criteria listed below.
Participants will receive a $50 stipend!
Criteria:
1. Are 18 years of age or older
2. Have been diagnosed with Acute Hepatic Porphyria (specific types include Acute Intermittent Porphyria, Variegate Porphyria, Hereditary Coproporphyria).
3. Own and are comfortable using a laptop or a desktop computer
4. Have internet access
Please call the APF at 866-APF-3635 to participate in the survey.
Volunteers Needed!
The APF will be exhibiting at the 2019 ACMG Annual Meeting in Seattle, WA April 3-5 AND Digestive Disease Week in San Diego, CA May 19-21. Are you interested in volunteering at our booth? If so, please contact Edrin Williams, Director of Patient Services, for additional information at edrinw@porphyriafoundation.org or 301.347.7166.
This is an incredible opportunity to raise awareness about porphyria to those with little knowledge about this rare disease.
RSVP Today
Patient Education and Support Meeting- Phoenix
LOCATION
Holiday Inn & Suites Phoenix Airport
DATE AND TIME
02/23/19 1:00pm - 02/23/19 3:00pm
3220 S. 48th Street Phoenix, AZ Room: Scottsdale Family and friends are welcome to attend!
Patient Education and Support Meeting- Ann Arbor
LOCATION
Holiday Inn
DATE AND TIME
03/16/19 1:00pm - 03/16/19 3:00pm
3600 Plymouth Road Ann Arbor, MI Room: Wolverine A Family and friends are welcome to attend!
Patient Education and Support Meeting- Seattle
LOCATION
Seattle Public Library- Central
DATE AND TIME
04/04/19 5:30pm - 04/04/19 7:30pm
1000 Fourth Avenue Seattle, WA Room: Washington Mutual Level: 4 Family and friends are welcome to attend!
Contact Information
Is your contact information up to date?
If not please give us a call @ 866-APF-3635 or Email to porphyrus@porphyriafoundation.com
American Porphyria Foundation 1.866.APF.3635 porphyriafoundation.org
STAY CONNECTED
"Remember.Research is the Key to Your Cure!"
Topic- How do patients with rare diseases experience the medical encounter? Exploring role behavior and its impact on patientphysician interaction
I enjoyed reading this so much and I think you will to. Topic- How do patients with rare diseases experience the medical encounter? Exploring role behavior and its impact on patientphysician interaction Author links open overlay panel Karolina BudychaThomas M.HelmsaCarstenSchultzb
How do patients with rare diseases experience the medical encounter?
Exploring role behavior and its impact on patientphysician interaction
Author links open overlay panel open access
Abstract
Objectives
Empirical research shows that patients with severe illnesses prefer the physician to dominate decision processes and provide the information needed. However, in rare diseases, due to the low prevalence and the lack of expertise, the patient is forced to become knowledgeable about his own disease state. Objectives of this study were to describe the experiences of patientphysician interaction in rare diseases, to develop an empirically derived typology of interaction patterns and to explore the antecedents of these interaction patterns, with a special focus on role behavior. Building on these results, implications for health care policy are made.
Methods
We designed an exploratory study as a series of semi-standardized interviews with patients suffering from rare diseases. We extracted the following six rare diseases: amyotrophic lateral sclerosis, Duchenne muscular dystrophy, epidermolysis bullosa, Marfan syndrome, neurodegeneration with brain iron accumulation and Wilson's disease. A total of 107 interviews were recorded, transcribed and analyzed thematically in accordance with the grounded theory tradition.
Results
As suggested, insufficient expertise of the healthcare providers proved to be a major problem in the highly specialized treatment process of rare diseases. Here, the patient often becomes an expert in his disease. Therefore, we identified the patient-directed interaction as a widely experienced communication pattern among patients with rare diseases. Our study also showed that role discrepancies have a major impact on communication processes in this context.
Conclusions
People with rare diseases often face challenges, due to the low prevalence and the resulting lack of knowledge of their healthcare providers. Communication processes in this context are mainly affected by the role behavior of both the patient and provider. The present study showed the relevance of the provider's ability to acknowledge the active role of the patient as an informed, involved and interactive partner in the treatment process. However, allowing the patient to control therapy may require a change of mind-set with some long-standing traditional roles in healthcare. Click to read more: https://www.sciencedirect.com/science/article/pii/S0168851012000644?fbclid=IwAR3-KkcpI2Ytt4J1swm9tFWg-JoyGyxAihpX8MNbxyEIf_Pz93-2r1cQIXQ
April 6-13, 2019. This is a chance for YOU to promote porphyria awareness and education within your community!!!
The APF will be providing tools for you to help you engage your community but we need YOU to participate and spread the word about porphyria. Stay tuned for more updates on this exciting week!
Attention Acute Porphyria membersYour help is needed!
A global research company is initiating an online survey to get a deeper understanding of patientsexperiences living with acute porhyria as well as understand how well a hypothetical new treatment would meet their needs. The study can be taken on a laptop or desktop computer and will take approximately 30 minutes to complete.
Participants will be screened to ensure they meet the criteria listed below, and those who complete the survey will receive a stipend.
Criteria:
1.Are 18 years of age or older
2.Have been diagnosed with Acute Hepatic Porphyria (specific types include Acute Intermittent Porphyria, Variegate Porphyria, Hereditary Coproporphyria).
3. Own and are comfortable using a laptop or a desktop computer
4.Have internet access
Please call the APF at 866-APF-3635 participate in the survey.
Volunteers Needed!
The APF will be exhibiting at the 2019 ACMG Annual Meeting in Seattle, WAApril 3-5. Are you interested in volunteering at our booth? If so, please contact Edrin Williams, Director of Patient Services, for additional information at edrinw@porphyriafoundation.org or 301.347.7166.
This is an incredible opportunity to raise awareness about porphyria to those with little knowledge about this rare disease.
Attention Shadow Jumpers
Shadow Jumpers, a graphic novel written by Shruti Shenbagam, Hetanshi Naik, and Dr. Manisha Balwani and illustrated by Tina Chu, is now available on the APF store for $15.
This book is designed specifically to educate children with EPP and their families in a way that everyone will understand and enjoy.
Get ready for FIND YOUR SHADOW 2019! Visit the APF Website for this year's application!
Once again this year, the Shadow Jumpers program will be selecting one family for an all-expenses paid trip to Walt Disney World in Orlando, FL. All children ages 5-17 and their families are eligible to apply. Shadow Jumpers is dedicated to finding creative ways to help kids and their families to take on a safe adventure.
In 2018, the Stuhlsatz family was selected for Light the Moment 2018, the theme of last year's trip. Their trip - from beginning to end - was "a once in a lifetime experience!"
Shadow Jumpers is excited for your submissions to FIND YOUR SHADOW 2019!
*please note: Shadow Jumpers is funded through donations and charitable contributions separate from the APF.
RSVP Today
Patient Education and Support Meeting- Phoenix
LOCATION Holiday Inn & Suites Phoenix Airport
DATE AND TIME 02/23/19 1:00pm - 02/23/19 3:00pm
3220 S. 48th Street Phoenix, AZ Room: Scottsdale Family and friends are welcome to attend!
Patient Education and Support Meeting- Ann Arbor
LOCATION Holiday Inn
DATE AND TIME 03/16/19 1:00pm - 03/16/19 3:00pm
3600 Plymouth Road Ann Arbor, MI Room: Wolverine A Family and friends are welcome to attend!
Patient Education and Support Meeting- Seattle
LOCATION Seattle Public Library- Central
DATE AND TIME 04/04/19 5:30pm - 04/04/19 7:30pm
1000 Fourth Avenue Seattle, WA Room: Washington Mutual Level: 4 Family and friends are welcome to attend!
I come from a Central Oregon town with the population of about 1100 people. I moved back here to Central Oregon, where I was raised after 40 years of being away. I took my grandson to a local ER in Redmond, OR (population about 26,000) after an accident where he hurt his foot which needed to be checked out. I was making small talk with him in the waiting room and told him of some new information about porphyria I had learned. A woman in the waiting room overheard our conversation. So, as it goes in small towns; we started talking. She told me that she had porphyria also.
She was called in to see the triage nurse but her husband remained and we visited. I exchanged contact information with him and we briefly traded stories. She came back in the room shortly. My first question after finding out what kind of porphyria she had (AIP), I asked her how she was diagnosed; as I knew of my own 'war stories' of finally finding the correct diagnosis.
Her story was similar to mine and other porphyria patients, being brushed off by many regular doctors who do/did not understand the cluster of symptoms or level of pain involved during a porphyria attack. If your symptoms don't exactly fit a text book illness model then it is all in your head been-there-done-that. I was diagnosed about a year ago after several Oregon doctors turned their back on me not wanting to deal with the local medical political climate. The few who didn't turn me away hung in there with me and saved my life.
My new friend (from the ER waiting room) was diagnosed about 14 to 16 years ago in Eugene, Oregon. She was deathly ill, hospitalized and was first diagnosed with Guillain-Barre syndrome, yet they still did not know exactly what was going on with her. Her doctors were all puzzled.
She had one doctor who used her personal three days off from work researching the internet on her behalf and wondered if she might have porphyria. This doctor did the porphyria testing and sure enough she nailed the correct diagnosis of AIP. Those kind of doctors are few and far between.
What I find amazing about our chance encounter in the local ER waiting room was that we both found out we were afflicted with this very rare disease in a sparsely populated area. One of us being among only approximately 1 in 100,000 porphyria patients affected in the US. It makes our chance meeting even more amazing
My personal take on just how rare porphyria is: there are probably more porphyria patients out there that could be identified if our doctors had more exposure in their training about the disease; and lab technicians were trained to collect the specimens and process the samples correctly. That is my nickel's worth
Porphyria Post
Wednesday - January 23, 2019 @ 06:30:00
Porphyria Post
Member Stories
Share your story! We want to hear from youâ?¦
In preparation for Porphyria Awareness Week (April 6-13), the American Porphyria Foundation invites you to share your story with the porphyria community. With permission, all stories will be featured on the APF Website. We will also feature select stories in our quarterly newsletter.
Stories should be about 500 2000 words and full of your personality. Make sure to include information about your specific porphyria type, your diagnostic journey and how this disease has impacted your life.
We will be sending out a small token of our appreciation to the first 5 submissions.
We look forward to receiving your stories. The APF office hears regular feedback that the stories listed on our site have helped them feel less isolated and work toward diagnosis. Thank you for having a hand in helping others through your stories and words.
Send your stories to Edrin at edrinw@porphyriafoundation.org
Volunteers Needed!
The APF will be exhibiting at the 2019 ACMG Annual Meeting in Seattle, WA April 3-5. Are you interested in volunteering at our booth? If so, please contact Edrin Williams, Director of Patient Services, for additional information at edrinw@porphyriafoundation.org or 301.347.7166.
This is an incredible opportunity to raise awareness about Porphyria to those with little knowledge about this rare disease.
Attention Shadow Jumpers
Shadow Jumpers, a graphic novel written by Shruti Shenbagam, Hetanshi Naik, and Dr. Manisha Balwani and illustrated by Tina Chu, is now available on the APF store for $15.
This book is designed specifically to educate children with EPP and their families in a way that everyone will understand and enjoy.
Get ready for FIND YOUR SHADOW 2019! Visit the APF Website for this year's application!
Once again this year, the Shadow Jumpers program will be selecting one family for an all-expenses paid trip to Walt Disney World in Orlando, FL. All children ages 5-17 and their families are eligible to apply. Shadow Jumpers is dedicated to finding creative ways to help kids and their families to take on a safe adventure.
In 2018, the Stuhlsatz family was selected for Light the Moment 2018, the theme of last year's trip. Their trip - from beginning to end - was "a once in a lifetime experience!"
Shadow Jumpers is excited for your submissions to FIND YOUR SHADOW 2019!
*please note: Shadow Jumpers is funded through donations and charitable contributions separate from the APF.
Panhematin Study - Participants Needed!
Do you have AIP, VP or HCP? Are you interested in participating in research? If so, we are recruiting volunteers for the Panhematin Study.
Our researchers need people to volunteer for the Panhematin study. Completing this study will help prevent insurance companies from not paying for this treatment.
The purpose of this study is to determine if Panhematin is safe and effective for prevention of acute attacks of porphyria.
â?¢ Can you predict when your next attack will happen?
â?¢ Are you currently receiving prophylactic heme treatment?
Please contact Edrin at the APF office (301.347.7166) if you are interested in participating.
Be a leader in Rare Disease
The EveryLife Foundation and Global Genes are partnering to bring you the RARE on the Road Tour. Learn, recognize, connect and discover within the rare disease community and spread the word about porphyria. The tour will be in Boston, MA, Birmingham, AL, Denver, CO, and Sioux Falls, SD. Visit https://raretour.org/ to learn more about this amazing opportunity!
EPP Clinical Trials: Volunteers Needed!
We are close, but we need YOU!
A clinical trial with an oral investigational drug intended to reduce phototoxicity in people living with EPP is underway.
Please note that a Phase 3 study is very unlikely if Phase 2 is not completed. To date, this drug has proven nontoxic and there have been essentially no dropouts in the Phase 2 study to date.
This is an oral drug which makes administration simple.
This study in adults will pave the way for a near-future pediatric trial. EPP is a rare disease and patients must participate in order to get the drug approved.
To date there is no public information on the availability of Scenesse, and if approved, two drugs on the market may lower their cost.
We will connect you with a Research Coordinator that will answer all your questions and concerns. We will work to make this process as easy as possible for you.
**Note: If you have not been contacted by a research coordinator please reach out to the APF office at 301.347.7166. **
Eligibility Criteria:
â?¢ Confirmed diagnosis of EPP
â?¢ Provide written informed consent to participate
â?¢ Be willing and able to travel to all study sites for scheduled visits
â?¢ 18 - 70 years of age at the time of screening.
Sites enrolling:
â?¢ Mount Sinai - New York City
â?¢ Wake Forest Baptist University - Winston Salem, NC
â?¢ University of Miami - Miami, FL
â?¢ University of Texas Medical Branch - Galveston, TX
â?¢ University of Utah - Salt Lake City, UT
â?¢ University of California San Francisco - San Francisco, CA
We need YOU! Please dig deep and consider being part of this changing moment for all with EPP.
Contact us here at the APF office to get in contact with a research coordinator.
301.347.7166 or 1.866.APF.635
porphyrus@porphyriafoundation.com
"RememberResearch is the key to YOUR cure!"
Each Step Toward Finding an Effective Treatment is Important!
Harvoni Study - PCT
Do you have PCT? Are you interested in participating in research? Do you have Hepatitis C? If you answered YES to these questions, this is for you.
We need YOU for a clinical trial!
The purpose of this clinical trial is to assess whether Harvoni alone is an effective therapy in active PCT patients with Chronic Hepatitis C.
Who can participate?
1. Adult patients with PCT who also have Hepatitis C
If you are interested in participating please contact Edrin Williams, Director of Patient Services at the APF office at 301.347.7166 for additional information.
RSVP Today
Patient Education and Support Meeting- Phoenix
LOCATION
Holiday Inn & Suites Phoenix Airport
DATE AND TIME
02/23/19 1:00pm - 02/23/19 3:00pm
3220 S. 48th Street Phoenix, AZ Room: Scottsdale Family and friends are welcome to attend!
Patient Education and Support Meeting- Ann Arbor
LOCATION
Holiday Inn
DATE AND TIME
03/16/19 1:00pm - 03/16/19 3:00pm
3600 Plymouth Road Ann Arbor, MI Room: Wolverine A Family and friends are welcome to attend!
Contact Information
Is your contact information up to date?
If not please give us a call @ 866-APF-3635 or Email to porphyrus@porphyriafoundation.com
American Porphyria Foundation 1.866.APF.3635 porphyriafoundation.org
STAY CONNECTED
"Remember.Research is the Key to Your Cure!"
The Diagnosis and Management of Erythropoietic Protoporphyria
Monday - January 21, 2019 @ 15:25:43
The Diagnosis and Management of Erythropoietic Protoporphyria
Porphyrias are a group of metabolic disorders resulting from enzymatic defects in the heme biosynthetic pathway. Erythropoietic protoporphyria is thought to be the second most common porphyria seen in clinical practice. It is, however, commonly under-recognized and can lead to both cutaneous manifestations as well as derangement in hepatic function in a minority of patients. This review summarizes the current understanding of this disorder. Different treatment options are discussed with the goal of preventing liver damage. The roles of liver and bone marrow transplantation are also addressed.
Porphyrias are a group of metabolic disorders caused by defects in the heme biosynthetic pathway. Heme is the critical prosthetic group for numerous hemoproteins such as hemoglobin, myoglobin, catalase, and microsomal cytochromes. The term porphyriais derived from the Greek word porphyra,which means purple.The biochemical hallmark of porphyrias is overproduction and, ultimately, overexcretion of compounds called porphyrins, which have a deep red or purple color. The first clinical reports of porphyria appeared in the late nineteenth century and described a patient with severe cutaneous photosensitivity and brown pigmentation of the bones, which is characteristic of congenital erythropoietic porphyria. Soon thereafter, a case of acute porphyria was described in a person addicted to drugs who had urine the color of port wine and later died after taking the hypnotic drug sulfonomethane.
Porphyrias can present clinically either with neurovisceral symptoms and signs such as abdominal pain, constipation, and weakness or with cutaneous symptoms and signs. In hereditary coproporphyria and variegate porphyria, patients may present with both types of symptoms; in the other forms of porphyria, patients manifest only one type of clinical presentation. When considering therapy for porphyrias, it is useful to classify the disorders into two major categories: acute/inducible porphyrias or chronic cutaneous porphyrias (Table 1). Regardless of the specific type of acute porphyria or associated enzymatic defect, all of the acute porphyrias produce similar neurovisceral manifestations and should be managed in a similar manner. Management of cutaneous porphyria, though more specific to the particular type of porphyria, also involves the application of several general principles.
Table 1
Classification and Major Features of Human Porphyrias
Clinical features
Disease
Primary enzymatic defect
Autosomal inheritance
Neurovisceral symptoms
Photosensitivity dermatosis
Acute/inducible pofphyiias
ALA-D deficiency porphyria
ALA dehydratase
Recessive
+
--
Acute intermittent porphyria
PBG deaminase
Dominant
+
--
Hereditary coproporphyria
Coproporphyrinogen oxidase
Dominant
+
+
Variegate porphyria
Protoporphyrinogen oxidase
Dominant
+
+
Chronic cutaneous porphyrias
Congenital erythropoietic
Uroporphyrinogen III cosynthase
Recessive
--
++
Hepatoerythropoietic porphyria
Uroporphyrinogen decarboxylase
Recessive
+/-
+
Porphyria cutanea tarda
Uroporphyrinogen decarboxylase
Dominant (acquired variant exists)
--
+
Protoporphyria
Ferrochelatase
Dominantâ?
--*
+
ALA=5-aminolevulinate; ALA-D=ALA dehydratase; PBG=porphobilinogen. ALA is the first intermediate in the heme biosynthetic pathway, and PBG is the second intermediate in the heme biosynthetic pathway.
*A neurovisceral syndrome reminiscent of those observed in acute porphyrias has been described in several patients with protoporphyria and hepatic failure around the time of orthotopic liver transplantation;
â? autosomal recessive inheritance has been described.
Erythropoietic protoporphyria (EPP) was first described by Magnus and associates in 1961.1 Bonkovsky and colleagues demonstrated that heme synthase or ferrochelatase (FECH), an inner mitochondrial enzyme, was deficient in this disorder (Figure 1).2 Since then, the gene for FECH has been localized to 18q21,3,4and approximately 120 disease-producing mutations have been described to date. The gene has 11 exons and a minimum size of 45 kb. From early studies of kindreds with EPP, most investigators concluded that the disease displayed autosomal dominant inheritance with low penetrance and an increased propensity to develop chronic liver disease,5 though autosomal recessive forms of EPP have also been described.6 The activity of ferrochelatase in clinically affected patients is approximately 1525% of normal activity. The parents of many patients are asymptomatic and phenotypically normal, though approximately one half demonstrate decreases in ferrochelatase activity. More recent familial studies have identified a single nucleotide polymorphism (IVS3-48C), which leads to a lower level of expression of the normal FECH gene allele.7 Patients develop clinical disease with less than 25% of normal FECH activity when a mutated allele is inherited along with the low-level-expressing, wild-type allele, which usually harbors the IVS3-48C polymorphism. One recent paper stressed the importance of large deletions in the FECH gene exons as an important cause of mutation-negativeEPP.8
FECH carries out the ultimate step in heme biosynthesis, namely the insertion of ferrous iron into protoporphyrin for the formation of heme. The only difference between heme and protoporphyrin is the insertion of an iron atom into the tetrapyrrole ring in heme. This insertion increases the stability of the structure and results in the loss of its fluorescent properties. In EPP, the major site of protoporphyrin overproduction is localized to the bone marrow, where erythrocytes are developed.912 Protoporphyrin, unlike other porphyrins, is water-insoluble and therefore removed from the body only through hepatic excretion into bile or feces. Protoporphyrin is not excreted in urine, though it does undergo some enterohepatic circulation.
Of all the porphyrias found in the United States, EPP is thought to be second in prevalence only to porphyria cutanea tarda. The exact prevalence of EPP has not been reliably estimated due to its frequently mild nature and normal urinary porphyrins, which increase the possibility of missing bona fide cases. There is no gender predominance in EPP, which is found across all ethnic and racial groups.
EPP, a disorder with highly variable clinical expression, has been previously well characterized as a chronic cutaneous porphyria.1316 Most affected patients present in early childhood with sun sensitivity and, at times, an urticarial-type reaction. Infants and children classically develop intense burning pain from sun-exposed skin following brief exposure in the spring and summer. Several hours later, erythema, edema, and itching become prominent. Vesicles are uncommon and develop only with prolonged sun exposure. With chronic and repeated exposure, involved skin may become leathery and hyperkeratotic. This reaction is particularly prominent in a malar butterflypattern on the face and over the knuckles of the hands. Patients usually have a chronic and stable disease course, with no identified aggravating factors.
Photoactivation of lipid-soluble protoporphyrin, which is deposited in the dermal blood vessels, leads to the characteristic skin burning from sun exposure. Chronic facial scarring, hirsutism, and fluorescent teeth, which are common in congenital erythropoietic porphyria or hepatoerythropoietic porphyria, are not usually found in EPP.16 Photoactivation of circulating water-soluble uroporphyrins, on the other hand, leads to chronic blistering, as seen in congenital erythropoietic porphyria and porphyria cutanea tarda.
The most serious complication of EPP is development of pigmentary cirrhosis of the liver.17,18 Fortunately, liver injury is evident in only a minority of patients with EPP (-10%), with approximately 35% of the patients eventually developing end-stage liver disease. The pattern of liver injury, when seen in this disorder, typically develops over a period of time and ranges from mild liver disease with elevations in liver enzymes to progressive liver disease with extensive bridging fibrosis or cirrhosis and other concomitant complications. By itself, EPP rarely presents as true acute liver injury, though there may be sudden acute worsening of underlying chronic liver disease.19 Liver disease is thought to develop due to the precipitation of protoporphyrin in hepatocytes and biliary radicles.20,21 No risk factors have been identified that are able to predict the development of liver disease in EPP thus far.
A common complication of EPP is the development of pigment gallstones, which have a high content of protoporphyrin. In bile, protoporphyrin may crystallize, providing the nidus for development of gallstones. When evident in children, gallstones should prompt evaluation for EPP.22 Mild anemia with or without hemolysis is occasionally noted. In EPP, abdominal pain and neuropathy-like symptoms are seen only rarely and in the setting of advanced liver disease.2326 Of note, motor neuropathy can develop both pre- and postoperatively at the time of liver transplantation.
The diagnosis of EPP requires the demonstration of increased levels of protoporphyrin, without increased levels of coproporphyrin, in the stool, RBCs, or both.27 In EPP, elevated levels of erythrocyte protoporphyrin, plasma protoporphyrin, and stool protoporphyrin are evident. Urinary porphyrin levels are usually normal because, as already mentioned, protoporphyrin is not excreted into the urine. Analysis of the plasma porphyrin emission spectrum reveals a characteristic peak at approximately 634636 nm, following excitation with light in the Soret band (-400 nm). Erythrocyte protoporphyrin elevations are also evident in severe iron deficiency and lead poisoning. In EPP, the protoporphyrin is primarily free protoporphyrin, in contrast to iron and lead toxicity, in which predominantly Zn-protoporphyrin is found.28In EPP patients, rising plasma protoporphyrin levels, along with severe elevations in RBC protoporphyrin levels, may portend impending liver failure.29,30 Patients with EPP who develop significant liver disease have been shown to have significant increases in urinary coproporphyrin levels. An inversion of the physiological urinary coproporphyrin isomer III/I ratio has also been observed in patients with protoporphyria who develop cholestatic cirrhosis.31 Liver biopsy is performed to assess the severity of suspected liver disease, particularly in patients with very high RBC (>5,000 µg/dL) or plasma protoporphyrin (>1,000 µg/dL) concentrations. Polarized microscopy of the biopsy may demonstrate birefringent crystal deposits with characteristic Maltese cross patterns in EPP. Plugging of biliary canaliculi with protoporphyrin may also be evident in patients with advanced liver disease.32
A multitude of treatment options are available for EPP,27 ranging from efforts to reduce protoporphyrin overproduction in the bone marrow to augmenting its excretion into bile (Table 2). Other agents are used for their cytoprotective properties in conjunction with measures to reduce the circulating pool of protoporphyrin and, on occasion, liver transplantation to correct end-organ damage. Cutaneous protection with opaque sunscreens and barrier clothing cannot be overemphasized. Standard sunscreens are not useful; the only topical sunscreens that are effective at blocking wavelengths greater than 400 nm, with a high sun protection factor (>30), are light-opaque, contain zinc oxide or titanium dioxide, and may be cosmetically unacceptable to some patients. Oral beta-carotene (Solatene, Roche; 60180 mg orally per day) reduces photosensitivity in approximately 80% of patients over a 13 month period after initiation of therapy. Beta-carotene causes yellow-orange discoloration of the skin, and topical beta-carotene cream is ineffective. Beta-carotene is thought to scavenge the free radicals generated by the excitation of protoporphyrin and is dosed to maintain a therapeutic serum level of 600800 pg/dL.33 Beta-carotene's efficacy has been questioned in the recent literature, and many patients have found their resulting yellow skin difficult to accept. Oral cysteine has also been used and is thought to have a similar mechanism of action.34 Bile-acid binding agents such as cholestyramine35,36 have been used to decrease the enterohepatic circulation of protoporphyrin. Patients have attempted to use long-term activated charcoal as a safe and cheap alternative in the hopes of decreasing intestinal reabsorption of protoporphyrin.37 Intravenous vitamin E was reported in a case report as being effective for reversing liver disease.38 Ursodeoxycholic acid has been used in patients with early liver disease,31 not only because of its ability to enhance the biliary excretion of protoporphyrin, but also because of its cytoprotective properties.3941 However, a recent study in an autosomal recessive mouse model of EPP demonstrated no benefit with the use of ursodeoxycholic acid and heme arginate.42 Patients are advised against caloric restriction and should undergo iron replacement only if they are found to be iron-deficient.4347
Table 2
Targets for Intervention
Increase protoporphyrin excretion into bile: ursodeoxycholic acid
Reduce protoporphyrin production: hematin and red blood cell transfusion
Decrease the circulating pool of protoporphyrin
Removal: plasmapheresis, hemodialysis, and exchange transfusions
Interrupting enterohepatic circulation: cholestyramine and activated charcoal
Use antioxidants or cytoprotective agents: vitamin E and ursodeoxycholic acid
Perform liver transplantation
Correct the underlying defect: bone marrow transplantation
Chronic therapy with intravenous hematin and erythrocyte transfusions is thought to suppress heme production4851 by decreasing protoporphyrin levels. Exchange transfusions and plasmapheresis have been used to remove the protoporphyrin in transit, as a bridge to liver transplantation or other more definitive treatments. Heme therapy, along with plasmapheresis, has been noted to stabilize patients with advanced liver disease.5254 Nonbiologic liver assist devices have been used recently in an attempt to decrease the morbidity associated with motor neuropathy, both in the pre- and postoperative period.55
Since the first liver transplantation for EPP-related liver disease in 1980, EPP patients with progressive liver disease have been transplanted with acceptable outcomes. Liver transplant recipients from US centers have been shown to have 1-, 5-, and 10-year survival rates of 85%, 69%, and 47%, respectively.56 These survival rates are similar to those for liver transplants performed for other indications. However, recurrent EPP was found in 65% (11/17) of patients who survived more than 2 months post-transplant. The high incidence of recurrent disease post-transplant is not surprising, as the transplant itself does not correct the primary cause and major source of protoporphyrin overproduction, which has been shown to be in the bone marrow.57 Of all the porphyrias, EPP has the best established indication for liver transplantation as a therapeutic option.58 The previous medical literature, as well as more recent reports, have demonstrated a benefit in EPP patients who receive a bone marrow transplant.59,60 Successful bone marrow transplantation with or without liver transplantation, depending upon the severity of the liver disease, is considered the definitive treatment for EPP.
Patients with EPP are at increased risk of developing chronic liver disease and should be vaccinated for hepatitis A and B (and possibly hepatitis E in the near future) in case they are found not to be immune. They should be counseled to avoid hepatotoxins such as alcohol because of the risk of accelerating their liver disease.61 EPP patients with no evident liver disease should be monitored with liver enzymes and porphyrin levels, both serum and RBC protoporphyrin, at least every 6 months, if not more frequently, to detect early signs of liver injury.62 Patients with known chronic liver disease should also be screened regularly for hepatocellular carcinoma.
Effective management of EPP involves the judicious use of all available treatment options to prevent disease progression and possibly achieve cure. Treatment is directed at minimizing complications from sun damage, monitoring for development of liver disease, and stabilizing cholestatic liver disease once it develops, in the hopes of a more definitive treatment such as bone marrow transplantation with or without liver transplantation. The initial experience with bone marrow transplantation is promising, though the opportune moment to intervene remains unclear.
1. Magnus IA, Jarrett A, Prankerd TA, Rimington C. Erythropoietic protoporphyria. A new porphyria syndrome with solar urticaria due to protoporphyrinaemia. Lancet. 1961;2:448451. [PubMed]
2. Bonkovsky HL, Bloomer JR, Ebert PS, Mahoney MJ. Heme synthetase deficiency in human protoporphyria. Demonstration of the defect in liver and cultured skin fibroblasts. J Clin Invest. 1975;56:11391148. [PMC free article] [PubMed]
3. Taketani S, Inazawa J, Nakahashi Y, Abe T, Tokunaga R. Structure of the human ferrochelatase gene. Exon/intron gene organization and location of the gene to chromosome 18. EurJ Biochem. 1992;205:217222. [PubMed]
4. Whitcombe DM, Carter NP, Albertson DG, Smith SJ, Rhodes DA, Cox TM. Assignment of the human ferrochelatase gene (FECH) and a locus for protoporphyria to chromosome 18q22. Genomics. 1991;11:11521154. [PubMed]
5. Sarkany RP, Alexander GJ, Cox TM. Recessive inheritance of erythropoietic protoporphyria with liver failure. Lancet. 1994;343:13941396. [PubMed]
6. Gouya L, Martin-Schmitt C, Robreau AM, Austerlitz F, Da Silva V, et al. Contribution of a common single-nucleotide polymorphism to the genetic predisposition for erythropoietic protoporphyria. Am J Hum Genet. 2006;78:214. [PMC free article] [PubMed]
7. Gouya L, Puy H, Robreau AM, Bourgeois M, Lamoril J, et al. The penetrance of dominant erythropoietic protoporphyria is modulated by expression of wildtype FECH. Nat Genet. 2002;30:2728.[PubMed]
8. Whatley SD, Mason NG, Holme SA, Anstey AV, Elder GH, Badminton MN. Gene dosage analysis identifies large deletions of the FECH gene in 10% of families with erythropoietic protoporphyria. J Invest Dermatol. 2007;127:27902794. [PubMed]
9. Straka JG, Hill HD, Krikava JM, Kools AM, Bloomer JR. Immunochemical studies of ferrochelatase protein: characterization of the normal and mutant protein in bovine and human protoporphyria. Am J Hum Genet. 1991;48:7278. [PMC free article] [PubMed]
10. Rufenacht UB, Gouya L, Schneider-Yin X, Puy H, Schäfer BW, et al. Systematic analysis of molecular defects in the ferrochelatase gene from patients with erythropoietic protoporphyria. Am J Hum Genet. 1998;62:13411352. [PMC free article] [PubMed]
11. Bloomer JR, Hill HD, Kools AM, Straka JG. Heme synthesis in protoporphyria. Curr Probl Dermatol. 1991;20:135147. [PubMed]
12. Samuel D, Boboc B, Bernuau J, Bismuth H, Benhamou JP. Liver transplantation for protoporphyria. Evidence for the predominant role of the erythropoietic tissue in protoporphyrin overproduction. Gastroenterology. 1988;95:816819. [PubMed]
13. Schmidt H, Snitker G, Thomsen K, Lintrup J. Erythropoietic protoporphyria. A clinical study based on 29 cases in 14 families. Arch Dermatol. 1974;110:5864. [PubMed]
14. DeLeo VA, Poh-Fitzpatrick M, Mathews-Roth M, Harber LC. Erythropoietic protoporphyria. 10 years experience. Am J Med. 1976;60:822. [PubMed]
15. Baart de la Faille H, Bijlmer-Iest JC, van Hattum J, Koningsberger J, Rade-makers LH, van Weelden H. Erythropoietic protoporphyria: clinical aspects with emphasis on the skin. Curr Probl Dermatol. 1991;20:123134. [PubMed]
16. Holme SA, Anstey AV, Finlay AY, Elder GH, Badminton MN. Erythropoietic protoporphyria in the U.K.: clinical features and effect on quality of life. Br J Dermatol. 2006;155:574581. [PubMed]
17. Chen FP, Risheg H, Liu Y, Bloomer J. Ferrochelatase gene mutations in erythropoietic protoporphyria: focus on liver disease. Cell Mol Biol (Noisy-le-grand) 2002;48:8389. [PubMed]
18. Poh-Fitzpatrick MB. The erythropoietic porphyrias. Dermatol Clin. 1986;4:291296. [PubMed]
19. Reisenauer AK, Soon SL, Lee KK, Hanifin JM. Erythropoietic protoporphyria presenting with liver failure in adulthood. Dermatology. 2005;210:7273. [PubMed]
20. Lee RG, Avner DL, Berenson MM. Structure-function relationships of protoporphyrin-induced liver injury. Arch Pathol Lab Med. 1984;108:744746. [PubMed]
22. Todd DJ. Gallstones in children. Am J Dis Child. 1991;145:971972. [PubMed]
23. Rank JM, Carithers R, Bloomer J. Evidence for neurological dysfunction in end-stage protoporphyric liver disease. Hepatology. 1993;18:14041409. [PubMed]
24. Muley SA, Midani HA, Rank JM, Carithers R, Parry GJ. Neuropathy in erythropoietic protoporphyrias. Neurology. 1998;51:262265. [PubMed]
25. Lock G, Holstege A, Mueller AR, Christe W, Doss MO, et al. Liver failure in erythropoietic protoporphyria associated with choledocholithiasis and severe posttransplantation polyneuropathy. Liver. 1996;16:211217. [PubMed]
26. Nguyen L, Blust M, Bailin M, Melendez L, Raines DE. Photosensitivity and perioperative polyneuropathy complicating orthotopic liver transplantation in a patient with erythropoietic protoporphyria. Anesthesiology. 1999;91:11731175. [PubMed]
27. Bonkovsky HL, Thapar M. Porphyria. In: Rakel RE, Bope ET, editors. Conn's Current Therapy.Philadelphia, PA: Elsevier Health; 2008. pp. 469474.
28. Lamola AA, Piomelli S, Poh-Fitzpatrick MG, Yamane T, Harber LC. Erythropoietic protoporphyria and lead intoxication: the molecular basis for difference in cutaneous photosensitivity. II. Different binding of erythrocyte protoporphyrin to hemoglobin. J Clin Invest. 1975;56:15281535. [PMC free article][PubMed]
29. Poh-Fitzpatrick MB. Protoporphyrin metabolic balance in human protoporphyria. Gastroenterology. 1985;88:12391242. [PubMed]
30. Bloomer JR. The liver in protoporphyria. Hepatology. 1988;8:402407. [PubMed]
31. Gross U, Frank M, Doss MO. Hepatic complications of erythropoietic protoporphyria. Photodermatol Photoimmunol Photomed. 1998;14:5257. [PubMed]
32. MacDonald DM, Germain D, Perrot H. The histopathology and ultrastructure of liver disease in erythropoietic protoporphyria. Br J Dermatol. 1981;104:717. [PubMed]
33. Mathews-Roth MM, Pathak MA, Fitzpatrick TB, Harber LH, Kass EH. Beta carotene therapy for erythropoietic protoporphyria and other photosensitivity diseases. Arch Dermatol. 1977;113:12291232.[PubMed]
34. Mathews-Roth MM, Rosner B. Long-term treatment of erythropoietic protoporphyria with cysteine. Photodermatol Photoimmunol Photomed. 2002;18:307309. [PubMed]
35. McCullough AJ, Barron D, Mullen KD, Petrelli M, Park MC, et al. Fecal protoporphyrin excretion in erythropoietic protoporphyria: effect of cholestyramine and bile acid feeding. Gastroenterology. 1988;94:177181. [PubMed]
36. Bloomer JR. Pathogenesis and therapy of liver disease in protoporphyria. Yale J Biol Med. 1979;52:3948. [PMC free article] [PubMed]
37. Gorchein A, Foster GR. Liver failure in protoporphyria: long-term treatment with oral charcoal. Hepatology. 1999;29:995996. [PubMed]
38. Komatsu H, Ishii K, Imamura K, Maruyama K, Yonei Y, et al. A case of erythropoietic protoporphyria with liver cirrhosis suggesting a therapeutic value of supplementation with alpha-tocopherol. Hepatol Res. 2000;18:298309. [PubMed]
39. Pirlich M, Lochs H, Schmidt HH. Liver cirrhosis in erythropoietic protoporphyria: improvement of liver function with ursodeoxycholic acid. Am J Gastroenterol. 2001;96:34683469. [PubMed]
40. Rademakers LH, Cleton MI, Kooijman C, Baart de la Faille H, van Hattum J. Early involvement of hepatic parenchymal cells in erythrohepatic protoporphyria? An ultrastructural study of patients with and without overt liver disease and the effect of chenodeoxycholic acid treatment. Hepatology. 1990;11:449457. [PubMed]
41. Van Hattum J, Baart de la Faille H, Van den Berg JW, Edixhoven-Bosdijk A, Wilson JH. Chenodeoxycholic acid therapy in erythrohepatic protoporphyria. J Hepatol. 1986;3:407412. [PubMed]
43. Holme SA, Worwood M, Anstey AV, Elder GH, Badminton MN. Erythropoiesis and iron metabolism in dominant erythropoietic protoporphyria. Blood. 2007;110:41084110. [PubMed]
44. Holme SA, Thomas CL, Whatley SD, Bentley DP, Anstey AV, Badminton MN. Symptomatic response of erythropoietic protoporphyria to iron supplementation. J Am Acad Dermatol. 2007;56:10701072.[PubMed]
45. Gordeuk VR, Brittenham GM, Hawkins CW, Mukhtar H, Bickers DR. Iron therapy for hepatic dysfunction in erythropoietic protoporphyria. Ann Intern Med. 1986;105:2731. [PubMed]
46. Milligan A, Graham-Brown RA, Sarkany I, Baker H. Erythropoietic protoporphyria exacerbated by oral iron therapy. Br J Dermatol. 1988;119:6366. [PubMed]
47. McClements BM, Bingham A, Callender ME, Trimble ER. Erythropoietic protoporphyria and iron therapy. Br J Dermatol. 1990;122:423424. [PubMed]
48. van Wijk HJ, van Hattum J, Baart de la Faille H, van den Berg JW, Edixhoven-Bosdijk A, Wilson JH. Blood exchange and transfusion therapy for acute cholestasis in protoporphyria. Dig Dis Sci. 1988;33:16211625. [PubMed]
49. Bloomer JR, Pierach CA. Effect of hematin administration to patients with protoporphyria and liver disease. Hepatology. 1982;2:817821. [PubMed]
50. Todd DJ, Callender ME, Mayne EE, Walsh M, Burrows D. Erythropoietic protoporphyria, transfusion therapy and liver disease. Br J Dermatol. 1992;127:534537. [PubMed]
51. Lamon JM, Poh-Fitzpatrick MB, Lamola AA. Hepatic protoporphyrin production in human protoporphyria. Effects of intravenous hematin and analysis of erythrocyte protoporphyrin distribution. Gastroenterology. 1980;79:115125. [PubMed]
52. Reichheld JH, Katz E, Banner BF, Szymanski IO, Saltzman JR, Bonkovsky HL. The value of intravenous heme-albumin and plasmapheresis in reducing postoperative complications of orthotopic liver transplantation for erythropoietic protoporphyria. Transplantation. 1999;67:922928. [PubMed]
53. Dellon ES, Szczepiorkowski ZM, Dzik WH, Graeme-Cook F, Ades A, et al. Treatment of recurrent allograft dysfunction with intravenous hematin after liver transplantation for erythropoietic protoporphyria. Transplantation. 2002;73:911915. [PubMed]
54. Do KD, Banner BF, Katz E, Szymanski IO, Bonkovsky HL. Benefits of chronic plasmapheresis and intravenous heme-albumin in erythropoietic protoporphyria after orthotopic liver transplantation. Transplantation. 2002;73:469472. [PubMed]
55. Eefsen M, Rasmussen A, Wulf HC, Brock A, Hansen BA. Erythropoietic protoporphyria and pretransplantation treatment with nonbiological liver assist devices. Liver Transpl. 2007;13:655657.[PubMed]
57. Poh-Fitzpatrick MB, Wang X, Anderson KE, Bloomer JR, Bolwell B, Lichtin AE. Erythropoietic protoporphyria: altered phenotype after bone marrow transplantation for myelogenous leukemia in a patient heteroallelic for ferrochelatase gene mutations. J Am Acad Dermatol. 2002;46:861866. [PubMed]
58. Seth AK, Badminton MN, Mirza D, Russell S, Elias E. Liver transplantation for porphyria: who, when, and how? Liver Transpl. 2007;13:12191227. [PubMed]
59. Wahlin S, Aschan J, Bjornstedt M, Broome U, Harper P. Curative bone marrow transplantation in erythropoietic protoporphyria after reversal of severe cholestasis. J Hepatol. 2007;46:174179. [PubMed]
60. Rand EB, Bunin N, Cochran W, Ruchelli E, Olthoff KM, Bloomer JR. Sequential liver and bone marrow transplantation for treatment of erythropoietic protoporphyria. Pediatrics. 2006;118:e1896e1899.[PubMed]
61. Bonkovsky HL, Schned AR. Fatal liver failure in protoporphyria. Synergism between ethanol excess and the genetic defect. Gastroenterology. 1986;90:191201. [PubMed]
62. Mathews-Roth MM. The consequences of not diagnosing erythropoietic protoporphyria. Arch Dermatol. 1980;116:407. [PubMed]
Articles from Gastroenterology & Hepatology are provided here courtesy of Millenium Medical Publishing
In preparation for Porphyria Awareness Week (April 6-13), the American Porphyria Foundation invites you to share your story with the porphyria community. With permission, all stories will be featured on the APF Website. We will also feature select stories in our quarterly newsletter.
Stories should be about 500 2000 words and full of your personality. Make sure to include information about your specific porphyria type, your diagnostic journey and how this disease has impacted your life.
We will be sending out a small token of our appreciation to the first 5 submissions.
We look forward to receiving your stories. The APF office hears regular feedback that the stories listed on our site have helped them feel less isolated and work toward diagnosis. Thank you for having a hand in helping others through your stories and words.
The APF will be exhibiting at the 2019 ACMG Annual Meeting in Seattle, WA April 3-5. Are you interested in volunteering at our booth? If so, please contact Edrin Williams, Director of Patient Services, for additional information at edrinw@porphyriafoundation.org or 301.347.7166.
This is an incredible opportunity to raise awareness about Porphyria to those with little knowledge about this rare disease.
FDA GRANTS
SCENESSE PRIORITY REVIEW!
Clinuvel Pharmaceuticals announced yesterday that Scenesse has been granted Priority Review by the FDA - the New Drug Application will be reviewed by July 8, 2019. Visit www.clinuvel.com for full information.
The APF and you, the patient community, have been relentless in pursuit of this treatment. Writing letters, making calls, signing petitions, sending photographs, attending meetings - it worked!
We will continue to focus on this until this treatment is approved and available to all EPP patients in the US.
ATTENTION SHADOW JUMPERS
Get ready for FIND YOUR SHADOW 2019! The application will be on the APF website starting this Friday, January 11th!
Once again this year, the Shadow Jumpers program will be selecting one family for an all-expenses paid trip to Walt Disney World in Orlando, FL. All children ages 5-17 and their families are eligible to apply. Shadow Jumpers is dedicated to finding creative ways to help kids and their families to take on a safe adventure.
In 2018, the Stuhlsatz family was selected for Light the Moment 2018, the theme of last year's trip. Their trip - from beginning to end - was "a once in a lifetime experience!"
Shadow Jumpers is excited for your submissions to FIND YOUR SHADOW 2019!
*please note: Shadow Jumpers is funded through donations and charitable contributions separate from the APF.
Panhematin Study - Participants Needed!
Do you have AIP, VP or HCP? Are you interested in participating in research? If so, we are recruiting volunteers for the Panhematin Study.
Our researchers need people to volunteer for the Panhematin study. Completing this study will help prevent insurance companies from not paying for this treatment.
The purpose of this study is to determine if Panhematin is safe and effective for prevention of acute attacks of porphyria.
·Can you predict when your next attack will happen?
·Are you currently receiving prophylactic heme treatment?
Please contact Edrin at the APF office (301.347.7166) if you are interested in participating.
EPP Clinical Trials: Volunteers Needed!
We are close, but we need YOU!
A clinical trial with an oral investigational drug intended to reduce phototoxicity in people living with EPP is underway.
Please note that a Phase 3 study is very unlikely if Phase 2 is not completed. To date, this drug has proven nontoxic and there have been essentially no dropouts in the Phase 2 study to date.
This is an oral drug which makes administration simple.
This study in adults will pave the way for a near-future pediatric trial. EPP is a rare disease and patients must participate in order to get the drug approved.
To date there is no public information on the availability of Scenesse, and if approved, two drugs on the market may lower their cost.
We will connect you with a Research Coordinator that will answer all your questions and concerns. We will work to make this process as easy as possible for you.
**Note: If you have not been contacted by a research coordinator please reach out to the APF office at 301.347.7166. **
Eligibility Criteria:
·Confirmed diagnosis of EPP
·Provide written informed consent to participate
·Be willing and able to travel to all study sites for scheduled visits
·18 - 70 years of age at the time of screening.
Sites enrolling:
·Mount Sinai - New York City
·Wake Forest Baptist University - Winston Salem, NC
·University of Miami - Miami, FL
·University of Texas Medical Branch - Galveston, TX
·University of Utah - Salt Lake City, UT
·University of California San Francisco - San Francisco, CA
We need YOU! Please dig deep and consider being part of this changing moment for all with EPP.
Contact us here at the APF office to get in contact with a research coordinator.
Each Step Toward Finding an Effective Treatment is Important!
Harvoni Study - PCT
Harvoni Study - PCT
Do you have PCT? Are you interested in participating in research? Do you have Hepatitis C? If you answered YES to these questions, this is for you.
We need YOU for a clinical trial!
The purpose of this clinical trial is to assess whether Harvoni alone is an effective therapy in active PCT patients with Chronic Hepatitis C.
Who can participate?
1.Adult patients with PCT who also have Hepatitis C
If you are interested in participating please contact Edrin Williams, Director of Patient Services at the APF office at 301.347.7166 for additional information.
RSVP Today
Patient Education and Support Meeting- Phoenix
LOCATION Holiday Inn & Suites Phoenix Airport
DATE AND TIME 02/23/19 1:00pm - 02/23/19 3:00pm
3220 S. 48th Street Phoenix, AZ Room: Scottsdale Family and friends are welcome to attend!
Patient Education and Support Meeting- Ann Arbor
LOCATION Holiday Inn
DATE AND TIME 03/16/19 1:00pm - 03/16/19 3:00pm
3600 Plymouth Road Ann Arbor, MI Room: Wolverine A Family and friends are welcome to attend!
Please visit this area again soon for upcoming CME opportunities.
The Recent Advances in Heme Biosynthesis and the Porphyrias, the eight major inborn errors of heme biosynthesis symposium, was held in January of 2018. This important three-day symposium focused on the most recent findings on the molecular biology and regulation of heme biosynthesis and the clinical features, pathophysiology, and current treatment of the acute hepatic and erythropoietic porphyrias. Day 1 was devoted to the clinical aspects and current treatments of each Porphyria. Days 2 and 3 focused on the latest research findings on heme biosynthesis and emerging molecular-based treatments for the porphyrias. Also, there was a special session for patients on the latest advances in the Porphyrias. The Symposium offered up to 25.5 CME Credits.
Telemedicine Consultations
If you need to consult with a Porphyria specialist and are unable to do so because of the distance to a Porphyria center, you now have the opportunity to consult electronically.
A Porphyria specialist and member of the APF Scientific Advisory Board, Dr. Karl Anderson, will be consulting with Porphyria patients via a telemedicine service from the General Clinical Research Center of the University of Texas Medical Branch at Galveston.
To access this service, you need to first locate a video teleconferencing facility in your community. These facilities are often found in libraries, businesses, universities and research centers. Unfortunately, neither the APF nor Dr. Anderson can provide you with a list of facilities with teleconferencing capabilities. Next, call Dr. Karl Anderson (409) 772-4661 to arrange for an appointment and to find out what information you might need to have ready prior to the telemedicine visit.
During the teleconference, Dr. Anderson will discuss your case with you just as he would during a regular office visit. This project was funded in part by a grant from the American Porphyria Foundation.
Physician Information Kit Available
Upon request, the APF will send a comprehensive physician education packet on the acute porphyrias to physicians. It includes educational materials on the diagnosis and management of the acute porphyrias; information on biochemical and genetic testing; Panhematin treatment for the acute porphyrias; and a recent article from the Annals of Internal Medicine on diagnosis and management of the acute porphyrias. Please call our office at (301) 347-7166 to order the kit.
In addition, the APF will also send information on PCT and EPP to physicians upon request. Please call our office at (301) 347-7166 to order the packet.
These packets are delivered via USPS, for physicians only.
Porphyria Slide Set
This PowerPoint slide set provides valuable information about porphyria, including discussion covering porphyria types, causes, diagnosis, and treatment protocols. It also includes vital information focusing on Panhematin (hemin for injection).
We know growing up with EPP has its challenges. You may have to cover up and look different outside or may be stuck indoors on those hot sunny days but whats important is to know that you arent alone out there. Many kids, just like you, are dealing with long days finding things to do to dodge the sun and ways to let loose at sun down. Here at Shadow Jumpers, weve developed a section for Kids to hear from people just like YOU! Check back here soon to read some amazing testimonial stories from kids in our EPP community. EPP may be hard, but its something we continue to tackle together.
I wear an oversized hoodie the hood covers the sides of my face. (Brady, age 12)
Cut thumbholes in your sleeves to keep your jacket or sweatshirt over your hands without sliding up. This works great for young kids! (anonymous)
Wear a big hat and walk on the shady side of the street! (Brenda, EPP)
I have a bag prepared with a long-sleeved shirt or light jacket, hat, gloves, and face cover (I use a bandanna, some use a buff) that I take with me on cloudy days so If it clears up Im covered and dont put myself in DANGER. (Rob, EPP)
Our son wears a zip-up lightweight jacket with a hood in the summer so that more he can unzip it to get more air and stay cooler.
Buy inexpensive sports gloves (for example, at Five and Below). We cut the tips off the fingers so that he can still manage things with his hands.
QUICK SHADE!:
Use a taller person as shade (Rob, EPP)
Walk next to buildings for shade (anonymous)
Tell teachers, coaches, other parents, etc. that THEY are your childs quickest protectionâ?¦just stand between them and the sun! (Kristen, caregiver)
Hold their hands to protect them from the sun especially when you dont have any gloves.
Always put your back to the sun.
SCHOOL:
Meet with teachers at the beginning of each year to explain EPP and offer guidelines for safety (caregiver)
Initiate a 504 plan for your child. (Ask the APF for a copy of a sample plan!)
Make sure your teachers know that your child needs a safe place for fire drills, recess, physical education class and field trips.
DRIVING:
Carry a blanket or towel in your car at all times and close it in the top of the window to provide shade. (Kristen, caregiver)
Have your car windows tinted with as dark as possible tinting that is allowed in your area.
VACATIONS:
We always bring along a canopy on vacation. We can set it up wherever our son is to provide him some safe shade. (EPP caregiver)
THINGS TO DO INSIDE:
So often during the summer I would go to the movies while all my friends were away during the day at camp. From the popcorn to the cool temperature to combat the hot summer days, I would spend hours at a time throughout the week at my local theater.The downside? So many movies meant so many tickets which meant so much money spent.Thats why, with this new thing called MOVIE PASS, going to the movies has been more affordable. For one flat monthly fee of $9.95, movie passer customers can see one movie a day each day for the month. To sign up and too see theaters in your area qualify go to https://www.moviepass.com/
THINGS TO AVOID:
Remember that water and snow reflect the sun!
Sun comes through windows, too.
UV INDEX APPS:
Hey Shadow Jumpers! Heres a cool tip I wanted to pass alongâ?¦ (Craig, EPP)
People with EPP often check the UV index every day. I know I regularly check the UV index for the day to know what I should wear or think about doing. So often people traditionally check the newspaper or kids rely on their parents. Today I wanted to share with you some great UV Apps!
Wolfram Sun Exposure Reference App
Based on your skin type, what SPF youre wearing and the UV forecast for your location, this comprehensive app can predict exactly how long you can stay in the sun before burning. Organizing a trip to the shore? Check out a five-day UV forecast displayed on a map, and find out what hours each day you should minimize sun exposure. ($0.99; available for iOS)
Ultraviolet ~ UV Index
Keep things simple with this cool tracker, which displays the current UV index in your area using a large, vibrantly colored circle. Blues and greens mean youre in the clear while reds and purples mean a dangerously high index. General sun safety advice will tip you off for when its time to put on a hat, apply sunscreen or avoid going outside altogether. Just the bare necessities for when youre bare at the beach. (Free; available for iOS)
EPAs SunWise UV Index
The U.S. Environmental Protection Agency designed this easy-to-navigate application, which delivers location-based UV index information. Ideal for plan-ahead types, the most useful feature is the color-coded hourly forecast that makes it easy to spot when the UV index is highest. (Free; available for iOS and Android)
(PLEASE NOTE: EPP reactions occur from visible light. UV Index applications and apps will ONLY indicate ultra-violet light, not the visible light range that affects EPP)
Stories
You are not alone! There are kids across the US who jump from shadow to shadow just like you! You can read all about them hereâ?¦
Hi Victor, I just read your story. So very sorry it took so long to receive a diagnosis. You have an amazing optimism in spite of it all. Your poor mom, to be called abusive. I hope and pray you will find a cure. God bless!
In case of any metabolic disorder, treatment is fo
Monday - January 28, 2019 @ 03:38:02
In case of any metabolic disorder, treatment is focused on tackling abdominal fat, high blood pressure, high blood sugar, and unhealthy cholesterol levels. Therefore, effective IV therapy with proper IV infusions can help to achieve the goal. Hope, IV therapy is helping you out from these conditions. Take care and thanks for sharing your experience.
This post contains huge information about travel.
Friday - February 1, 2019 @ 12:26:11
This post contains huge information about travel. Useful post and informative content. Thank you so much for sharing the valuable post. For More Information Visit Here: Guy Sands-Pingot
IV hydration can be a safest and healthy therapy w
Thursday - February 7, 2019 @ 07:02:26
IV hydration can be a safest and healthy therapy which can turn your bachelor/bachelorette party into a beast. you may be well aware that you will need recovery relief from the activities partaken in during the bachelor or bachelorette weekend. IV Therapy in Miami can make your bachelor or bachelorette weekend more smooth and funny.
Such a wonderful article in which clearly expresse
Saturday - February 9, 2019 @ 15:31:37
Such a wonderful article in which clearly expressed about safety. it is more informative and best article.We also provide best professional training courses online you can visit our website for more information and affordable training courses Professional Online Safety Training Courses.
I think this is an informative post and it is very
Monday - February 11, 2019 @ 04:33:02
I think this is an informative post and it is very useful and knowledgeable. therefore, I would like to thank you for the efforts you have made in writing this article. easyjet overbooking
I admire this article for the well-researched cont
Good to know about these services. Get constructio
Friday - March 1, 2019 @ 01:15:18
Good to know about these services. Get construction course of Fall Protection Online from Safety First Training Ltd. to work safely at the construction site.
My two sons have EPP. We do not live, unfortunatel
Wednesday - March 6, 2019 @ 08:05:20
My two sons have EPP. We do not live, unfortunately near a specialist. Not one time that they have had an outbreak has a doctor given them anything for pain! Once when one of my sons was burning so bad a nurse practioner suggested I give him percocet, so she wrote a whopping 6 pills. This helped alot but wasn't near enough. They only need something perhaps 2 or 3 times a year. Not on a regular basis. When they go to the doctor's office, clinic or emergency room they are treated like drug seekers! They have never been on legal or illegal drugs. It's a horrible shame this drug epidemic does not allow people with real pain some relief. As a mother, it kills me to see my children suffer. Someone PLEASE help!
Thanks for sharing the post with us. Home care ser
Saturday - March 9, 2019 @ 06:33:25
Thanks for sharing the post with us. Home care services are needed for the senior citizens and patients. Private Home Care St Louis Mo who is professional enough in providing the home care services.
i love reading this article so beautiful!!great jo
Good to know about this. Buy mens overcoat london from MALFORD OF LONDON which is the right destination to get the high-quality Bernard Weatherill tweed jacket online.









 Facebook Live with Dr. Bruce Wang
Facebook Live with Dr. Bruce Wang

 We need your help! The APF has created an extensive list of physicians around the world who are treating with people with Porphyria. This list helps when people from around the world contact the APF needing assistance finding a knowledgeable physician. YOU can help patients everywhere by sharing with us the name and location of your physician. Comment on this post, PM Edrin Williams, APF Director of Patient Services or call the APF office at 301.347.7166 to give us their information. Thank you!
We need your help! The APF has created an extensive list of physicians around the world who are treating with people with Porphyria. This list helps when people from around the world contact the APF needing assistance finding a knowledgeable physician. YOU can help patients everywhere by sharing with us the name and location of your physician. Comment on this post, PM Edrin Williams, APF Director of Patient Services or call the APF office at 301.347.7166 to give us their information. Thank you! The APF asked our Facebook friends for their top questions they would ask a porphyria expert.
The APF asked our Facebook friends for their top questions they would ask a porphyria expert.
 Dr. Bruce Wang, Porphyria Expert at UCSF and American Porphyria Foundation Protect the Future physician, has won Best Presentation at ICPP for his excellent work titled Single Cell Transcriptomic Analysis of Hepatocytes in a Mouse Model of Porphyria Cutanea Tardain collaboration with B. Patkar, D. Burhan, and J. Phillips. 118 abstracts from 27 countries were submitted for presentations and posters. Congratulations Dr. Wang!
Dr. Bruce Wang, Porphyria Expert at UCSF and American Porphyria Foundation Protect the Future physician, has won Best Presentation at ICPP for his excellent work titled Single Cell Transcriptomic Analysis of Hepatocytes in a Mouse Model of Porphyria Cutanea Tardain collaboration with B. Patkar, D. Burhan, and J. Phillips. 118 abstracts from 27 countries were submitted for presentations and posters. Congratulations Dr. Wang!  GPAC has been formalized. The APF is proud to be a founding member of the Global Porphyria Advocacy Coalition, an umbrella organization for porphyria patient advocacy groups worldwide. Work will focus on access to treatment, support, research, awareness and education of all the porphyrias. We are stronger together!
GPAC has been formalized. The APF is proud to be a founding member of the Global Porphyria Advocacy Coalition, an umbrella organization for porphyria patient advocacy groups worldwide. Work will focus on access to treatment, support, research, awareness and education of all the porphyrias. We are stronger together!
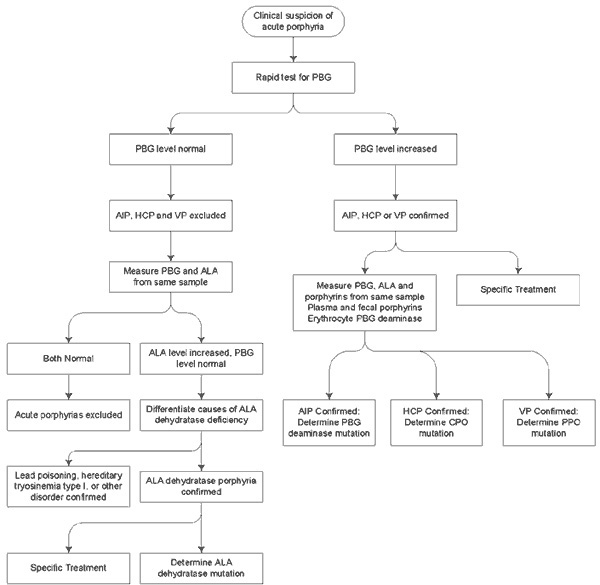
 Urine may appear purple during an attack or after standing in the light
Urine may appear purple during an attack or after standing in the light Medical Moment: Patient/Physician Relationship
Medical Moment: Patient/Physician Relationship

 Mitchell Felts, age 12
Mitchell Felts, age 12


 Nora began having difficulty in the sun during the summer when she was three. After being outside, she would cry inconsolably and in pain, sometimes for hours. The backs of her hands in particular seemed to bother her, and she would tell us they "itched." However, there were no outward manifestations-no rash, no swelling, no marks. We were stumped, as were the doctors we visited. At their recommendation, we tried various solutions, including Sarna lotion, Claritin, and weaning on and off various sunscreens-none of these worked. As a last resort, a dermatologist prescribed Benadryl, which worked only because it knocked her out.
Nora began having difficulty in the sun during the summer when she was three. After being outside, she would cry inconsolably and in pain, sometimes for hours. The backs of her hands in particular seemed to bother her, and she would tell us they "itched." However, there were no outward manifestations-no rash, no swelling, no marks. We were stumped, as were the doctors we visited. At their recommendation, we tried various solutions, including Sarna lotion, Claritin, and weaning on and off various sunscreens-none of these worked. As a last resort, a dermatologist prescribed Benadryl, which worked only because it knocked her out.
 ð??·To start giving to the APF click here. Under the Amazon search bar you will see the word SUPPORTING in orange, change the organization to reflect the American Porphyria Foundation. Then, you are ready to start shopping and supporting! By simply buying your normal amazon purchases through AmazonSmile you are impacting those affected by porphyria.
ð??·To start giving to the APF click here. Under the Amazon search bar you will see the word SUPPORTING in orange, change the organization to reflect the American Porphyria Foundation. Then, you are ready to start shopping and supporting! By simply buying your normal amazon purchases through AmazonSmile you are impacting those affected by porphyria. Here is our story on my son, Briggs. I am beyond grateful for the APF. Without the APF we may not be where we are today with a diagnosis.
Here is our story on my son, Briggs. I am beyond grateful for the APF. Without the APF we may not be where we are today with a diagnosis.















 MEET MILTON CUBAS, APF Member and ALAD Porphyria (ADP) patient.
MEET MILTON CUBAS, APF Member and ALAD Porphyria (ADP) patient. My name is Abdul Waheed Butt. I lived in Pakistan I am facing skin problem name Congenital Erythropoietic Porphyria (Gunther Disease) CEP. When I was born, I was completely normal like other children. When I was 2-months-old, my mother cut my hand nails there are starting water from it. When the nails water touch my face it becomes Water Blisters on my whole face. My parents checked me from so many Doctors in Pakistan, but they have no idea or treatment how to solve that problem. At-last, we found a skin specialist Dr. Ishfaq Ahmed who said that the disease name is CEP. He advised me to avoid sunlight and covered when I go out. The only treatment of this disease is to avoid sunlight. Then my parents covered me all the time for my safety as they can.
My name is Abdul Waheed Butt. I lived in Pakistan I am facing skin problem name Congenital Erythropoietic Porphyria (Gunther Disease) CEP. When I was born, I was completely normal like other children. When I was 2-months-old, my mother cut my hand nails there are starting water from it. When the nails water touch my face it becomes Water Blisters on my whole face. My parents checked me from so many Doctors in Pakistan, but they have no idea or treatment how to solve that problem. At-last, we found a skin specialist Dr. Ishfaq Ahmed who said that the disease name is CEP. He advised me to avoid sunlight and covered when I go out. The only treatment of this disease is to avoid sunlight. Then my parents covered me all the time for my safety as they can. My name is Cassandra King. There are three main things that define me: I am the daughter of the King of Kings (saved by grace through Jesus), I am the wife of Russ King, and I am the mama of Joshua King. There are, of course, other hats I wear: Im a daughter, a sister, an auntie, a friend, a volunteer, etc. And though, I really loathe to admit it, there is one other thing that defines me pain. Constant, unrelenting, heart-breaking pain. A broken body.
My name is Cassandra King. There are three main things that define me: I am the daughter of the King of Kings (saved by grace through Jesus), I am the wife of Russ King, and I am the mama of Joshua King. There are, of course, other hats I wear: Im a daughter, a sister, an auntie, a friend, a volunteer, etc. And though, I really loathe to admit it, there is one other thing that defines me pain. Constant, unrelenting, heart-breaking pain. A broken body. Now, Im kinda stubborn and Id try to outrun these attacks. But once the motor neuropathy would begin, Id cave quickly to going into the hospital for treatment. Russ became the pastor of our church in 2008, and I had the blessing of continuing as the church secretary for him for 2 more years. But, as I was still having these cyclical attacks, it left Russ and his staff under further pressure when I was out for health because they had to do my job as well as their own. And after 3 years of all these attacks, my body was not bouncing back after the attacks. My body was permanently damaged. So, I had to come to the hard decision to medically retirequit my job, that I loved deeply. I had no child at home, I was 26 years old, and I was unable to work because of my health.
Now, Im kinda stubborn and Id try to outrun these attacks. But once the motor neuropathy would begin, Id cave quickly to going into the hospital for treatment. Russ became the pastor of our church in 2008, and I had the blessing of continuing as the church secretary for him for 2 more years. But, as I was still having these cyclical attacks, it left Russ and his staff under further pressure when I was out for health because they had to do my job as well as their own. And after 3 years of all these attacks, my body was not bouncing back after the attacks. My body was permanently damaged. So, I had to come to the hard decision to medically retirequit my job, that I loved deeply. I had no child at home, I was 26 years old, and I was unable to work because of my health. I consider myself to be a pretty confident, healthy, and strong woman with a whole lot of life to live. Life is beautiful and I never knew how much I took it for granted until 2016 hit me. I didnt feel like myself at all. I was the heaviest I had ever been and couldnt muster up the energy to get through the day. Juggling a full-time job along with a side hustle teaching fitness classes, I thought certainly I am doing all of the right things and leading a healthy lifestyle, what could possibly be wrong?
I consider myself to be a pretty confident, healthy, and strong woman with a whole lot of life to live. Life is beautiful and I never knew how much I took it for granted until 2016 hit me. I didnt feel like myself at all. I was the heaviest I had ever been and couldnt muster up the energy to get through the day. Juggling a full-time job along with a side hustle teaching fitness classes, I thought certainly I am doing all of the right things and leading a healthy lifestyle, what could possibly be wrong? My name is Allison Linner and I am seventeen. I came into this world on a cold, rainy day in early October, but my Dad said that the moment I was born, the sun peeked through the clouds, thus my nickname, Sunshine.
My name is Allison Linner and I am seventeen. I came into this world on a cold, rainy day in early October, but my Dad said that the moment I was born, the sun peeked through the clouds, thus my nickname, Sunshine. My name is Cassidy Clark and I have Erythropoietic Protoporphyria (EPP).
My name is Cassidy Clark and I have Erythropoietic Protoporphyria (EPP).
 Please get your APF Merchandise today all sizes have been updated in the line item description. Prices are now on clearance. If you see a zero in the size you want we will NOT be ORDERING and ask that you choose another item. Watch out in the next week for the new tshirts-umbrellas-super nice-APF bags.
Please get your APF Merchandise today all sizes have been updated in the line item description. Prices are now on clearance. If you see a zero in the size you want we will NOT be ORDERING and ask that you choose another item. Watch out in the next week for the new tshirts-umbrellas-super nice-APF bags. 



 A research company has initiated an online survey to get a deeper understanding of patientsexperiences living with acute porphyria. The study can be taken on a laptop or desktop computer and will take approximately 30 minutes to complete. Participants will be screened to ensure they meet the criteria listed below.
A research company has initiated an online survey to get a deeper understanding of patientsexperiences living with acute porphyria. The study can be taken on a laptop or desktop computer and will take approximately 30 minutes to complete. Participants will be screened to ensure they meet the criteria listed below.

 We know growing up with EPP has its challenges. You may have to cover up and look different outside or may be stuck indoors on those hot sunny days but whats important is to know that you arent alone out there. Many kids, just like you, are dealing with long days finding things to do to dodge the sun and ways to let loose at sun down. Here at Shadow Jumpers, weve developed a section for Kids to hear from people just like YOU! Check back here soon to read some amazing testimonial stories from kids in our EPP community. EPP may be hard, but its something we continue to tackle together.
We know growing up with EPP has its challenges. You may have to cover up and look different outside or may be stuck indoors on those hot sunny days but whats important is to know that you arent alone out there. Many kids, just like you, are dealing with long days finding things to do to dodge the sun and ways to let loose at sun down. Here at Shadow Jumpers, weve developed a section for Kids to hear from people just like YOU! Check back here soon to read some amazing testimonial stories from kids in our EPP community. EPP may be hard, but its something we continue to tackle together.














 Facebook
Facebook Twitter
Twitter Google+
Google+



{kind=link}
{kind=link}