3:30 pm Research Overview and Patient Perspective TBD and Sharon Dill, Patient Representative
3:45 pm Q & A moderated by Co-Chairs
4:30 pm Closing remarks American Porphyria Foundation
Hyatt Regency Orlando
9801 International Drive
Orlando, Florida 32819
USA
+1 (407) 2841234
PSA- Urgent Need for Acute Porphyria Patients Research
Monday - December 18, 2017 @ 07:00:46
Dear friends,
Please consider being a volunteer. The new drug , Givosarin has had great results thus far..Stopping Attacks is the objective. Ppl like, Michael Boone and Lakeisha Johnson have been telling you all about the great experience.
Your travel is all paid! Miami, Winston Salem, Boston, Galveston beach, San Francisco, Salt Lake City. , Birmingham, NYC, Philadelphia, Little Rockoff you go..Meet an expert!
Please we urge all of you to contact the APF @
1-866-APF-3635 ask for Edrin!
Get involved with Research today!
Part 2- Helping Patients Take Charge of Their Chronic Illnesses
Monday - December 18, 2017 @ 06:00:52
Empowerment through education
It's very difficult for patients to do what they don't understand, so the first step in equipping patients to take on a more active role in their health care is to educate them. Start by communicating to patients that education is perhaps as important to their health as getting their prescriptions filled. They need to know all they can about their disease.
But just as patient-centered care can be more effective, patient-centered education is better education. The old education program, where you bring people in, sit them down and lecture to them, doesn't work any better than bringing them in, sitting them down and telling them to lose 20 pounds. Instead, the patient's needs should drive the education. For example, our center is testing diabetes education courses based entirely on questions from the audience. We do have a checklist of topics we want to cover, but we address those topics in the context of patient questions rather than through an impersonal lecture. Patients aren't interested in their disease from an intellectual perspective, as we are. They want to know about themselves. What does this mean to me? How's this different for me? How's it going to affect my life?
Four of the most important lessons patients with chronic diseases need to understand are the following:
1.Their illness is serious. There are still patients out there who believe they have the not-so-serious kind of diabetes. If they don't believe it is a problem, they will never make changes to improve their health.
2.Their condition is essentially self-managed. Every decision patients make throughout the day, from what they eat to whether they walk or ride the bus, has an influence on their health. Communicate to patients that they are the most important individuals in managing their illnesses.
3.They have options. There is rarely one perfect way to treat a condition. In the case of diabetes, for example, patients can be treated through diet and exercise, oral medication, insulin and so on. Patients need to understand the different treatment options available and should be encouraged to look at the personal costs and benefits of each. Only the patient can decide if the benefits are greater than the costs.
4.They can change their behavior. Rarely do patients leave the doctor's office and immediately enact whatever change was recommended. The reality is that it often has to be spread out into a series of steps. Teach patients that significant behavioral changes can be made by setting goals, taking that first step and figuring out what you learn about yourself along the way.
Empowering patients with information
One way to help patients focus and begin thinking about their health care goals is to talk with them about their individual health measures (e.g., blood pressure, LDL, HbA1c) and what those numbers mean. At our center, for example, we give patients a handout that lists the critical measures for their condition (ideal and actual), explains what those numbers mean and offers strategies for improvement. When faced with this information, patients can see for themselves where they are struggling and what they can do to better their scores.
A sample page from our handout is shown here.
Blood Pressure
Actual: _________________ mm/Hg
Ideal: 130/85 or lower
My goal is: ______________
A blood pressure reading has two numbers. The top number is called systolic blood pressure. This is the amount of pressure against the blood vessel walls when your heart pumps. The bottom number is called diastolic blood pressure. This is the amount of pressure against the blood vessel walls when your heart relaxes, that is, between heart beats.
In general, high blood pressure means that systolic blood pressure, diastolic blood pressure or both may be too high. For people with diabetes, high blood pressure is 130/85 or higher. High blood pressure increases your risk for strokes, heart attacks, kidney damage and eye disease.
To lower your blood pressure you can:
·Eat less salt,
·Take blood pressure medicine,
·Exercise,
·Stop smoking,
·Monitor blood pressure,
·Drink less alcohol,
·Maintain reasonable weight,
·Other: _________________________________
Helping patients set goals
In the patient-centered model of care, the driving force behind each patient visit is the patient's agenda or goals related to his or her condition. Ideally, the goal is clearly displayed in the patient's chart, and each person who handles the chart plays a part in supporting the patient in that goal, asking, How did it go? What have you done this week? How can we help you do better?
You might be thinking, My patients don't have goals, but they do. Even noncom-pliant patients have goals. Probably the best definition of noncompliance is a doctor and patient working toward different goals.
The process of setting self-management goals with the patient involves essentially two steps.
1.Start at the problem. Rather than beginning the patient encounter focused on lab values or weight or blood pressure readings, begin by saying, Tell me what concerns you most. Tell me what is hardest for you. Tell me what you're most distressed about and what you'd most like to change. You'll get to the lab values and other issues later, but it will be in the context of the patient's personal goal, which will make it more meaningful for the patient.
As you begin to get a sense of the patient's concerns, explore those issues together. Ask, Is there an underlying problem? Do you really want this problem to be solved? What's the realissue?
2.Develop a collaborative goal. Once you have worked with the patient to identify the real problem, your instinct may be to try to solve it, but don't. Don't try to fix it. Don't just say, It will be OK. Instead, validate the patient's feelings and his or her capacity to deal with the problem, and continue asking questions that will lead the patient to his or her own solution. Ask, What do you think would work? What have you tried in the past? What would you like to try?
It's always more meaningful when patients find the ah ha! on their own, so give them that chance. Encourage them to come up with ideas first, then offer your own suggestions or additional information that they may need. You can say this works for some people or have you tried this? or here's why I don't think that's a good idea. The important thing is to give the patient the opportunity to say no and to make the final decision on what goal to try.
Ultimately, at the end of the conversation, the patient should be able to tell you one step he or she is going to take. It should be very specific. If the patient says, I'm going to exercise more, ask what that means. Will they exercise four times a week? What activity will they be doing? How far will they walk? Help them to come up with a specific plan that they have created for themselves. It may not be the ultimate goal you would have chosen for the patient, but it's one they are more likely to accomplish. At the next visit, then, you can build on that.
Who actually works with patients to set their goals, whether you or the nurse or the diabetes educator, is perhaps less important than the fact that patients are encouraged to be more involved. The emphasis on self-management goals suggests that the visit is for them. It is their agenda, and they are active participants in the outcome.
Series overview
This article is part of an FPM series that followed Family Care Network, a northwest Washington state group without walls, as it tackled a 13-month quality improvement project focused on chronic disease care. The project was headed by the Institute for Healthcare Improvement and involved approximately 30 organizations nationwide.
The patient-centered model of care, and the emphasis on empowering patients to find their own solutions, may go against your instincts as a physician. As health care professionals, we often feel most helpful when we've given advice. The truth is, however, that we don't really help people solve their problems or make lasting changes in their lives by telling them what they should do. Ultimately, patients need to find their own solutions and motivation and must take responsibility for their health. We must empower them to do just that.
Martha Funnell, a certified diabetes educator, is the director for administration at the Michigan Diabetes Research and Training Center, University of Michigan Health System at Ann Arbor. The MDRTC has been funded by the National Institutes of Health since 1977 and is one of six such centers in the United States.
REFERENCE
1. Wagner EH, Austin BT, Von Koroff M. Improving outcomes in chronic illness. Managed Care Quarterly.1996;4(2):1225.
Light The Moment 2018
Friday - December 15, 2017 @ 07:00:20
INTRODUCING
A new program supported by the American Porphyria Foundation
for children living with Erythropoietic Protoporphyria (EPP)
Inside you will find tips and tricks on living with EPP, learn from fellow EPPers, read EPP Kid's stories, see new apparel and a very special opportunity withLIGHT THE MOMENT 2018!
For comprehensive information on Erythropoietic Protoporhyria (EPP), please visit the American Porphyria Foundation website: www.porphyriafoundation.org
Part 1- Helping Patients Take Charge of Their Chronic Illnesses
Thursday - December 14, 2017 @ 06:00:12
Helping Patients Take Charge of Their Chronic Illnesses
The best thing you can do for your patients with chronic diseases is to let them run with the ball.
Martha M. Funnell, MS, RN
Fam Pract Manag. 2000 Mar;7(3):47-51.
Noncompliance has always been a significant challenge in chronic disease care, and it's one of the issues Family Care Network has had to confront as part of its diabetes quality improvement project, which FPM is following (see Series overview). This article is derived from a presentation the author made to the physicians and staff members of Family Care Network as part of that project. As you'll see, the author's solution to noncompliance is simply to render it obsolete.
It's been said that, as health care professionals, we take credit for our patients' successes and blame them for our failures. How often have you heard a colleague say something like, I got Mrs. Smith's HbA1c down to 7 percent, but Mr. Jones was noncompliant. The truth is that, in both cases, the responsibility is shared, but ultimately the patient has more control of the outcome than we do.
Think about two of your patients who have, say, type-2 diabetes. Focus on one patient whose condition is well managed and another whose condition is not well managed. What would you say are the differences between those two? Why is one more successful at controlling the disease than the other? Very often, if you examine it closely, the key difference is the patient's level of involvement and responsibility for his or her own condition.
KEY POINTS:
·The old models of care, where physicians tell patients what to do and try to motivate them to change, do not work.
·Because patients' day-to-day decisions have a tremendous impact on their health, they must be active, informed participants in the health care process.
·Physicians can help patients take charge of their conditions by encouraging them to set self-management goals.
The old models
Our health care systems are not always designed with the patient in mind and do little to empower patients to take charge of their conditions. Instead, they are based on an old acute-care model, where the patient presents to his or her physician and the physician tells the patient what to do to get better. It is a do as I say model of care, and the patient's role is fairly passive.
That model works fine for a good deal of acute care and in some other circumstances, but it doesn't work well for chronic illness. Having recognized this, many health care systems have tried a different model in which the focus is compliance, or adherence. This model does a better job of recognizing that the patient needs to do something, but it doesn't give the patient any more power. The health care professional is still very much the authority trying to get the patient to do what is needed; the patient's job is simply to be obedient. What we've found, however, is that you can't get patients to do anything. The motivation to change one's behavior even to take one's medication is largely internal. The patient is responsible and must take an active role in his or her own care.
Of course the idea of an active patient can be taken to the extreme. Some patients, in response to the old models of care that haven't been working for them, have become not just active patients but activists. They come to their visits with (mis)information from the Internet and other sources; and, although they are involved in their care, they are involved in an adversarial way. This doesn't create better outcomes, and it can result in an unproductive doctor-patient relationship.
The failure of the traditional models has led the health care community to ask, what kind of approach can we use to deal with chronic illness, recognizing that our influence over patients' behavior is limited and fairly temporary? In other words, how can we change chronic illness care so that it better fits with chronic illness?
The patient is the solution
We have learned over the years at our center that effective chronic illness care requires two things. First, it requires a team with the patient at the center. Second, it requires active, involved participants especially an active, involved patient. This model of care can be described using various terms empowerment, informed choice, patient centered but they all have the same underlying concept: The patient is at the center and is actively involved in his or her own health care.
Then try walking after dinner every night with your husband for 10 minutes.
What do you hate about it? What would help you do better at it?
I don't think I can quit smoking.
Smoking is the leading cause of preventable death
Why do you think that? What has happened in the past when you tried to quit? What concerns you most when you think about trying to quit?
I haven't been able to test my blood sugar four times a day.
It's hard at first, but just keep trying. You really need to keep track of it.
What is preventing you from doing that? Do you know what the numbers mean?
But why can't we stick with the old models? Why does the patient need to be so involved? There are several reasons.
First, most chronic illness care does not even involve physicians and other health care professionals. Instead, it's estimated that between 95 percent and 99 percent of chronic illness care is given by the person who has the illness. On a day-to-day basis, the patient is in charge of his or her own health, and the daily decisions people make have a huge impact on patient outcomes and quality of life.
Second, as a family physician, you may know what's best for treating diabetes or asthma or congestive heart failure, but that does not mean you necessarily know what's best for an individual patient. Even in close doctor-patient relationships, you can't always know the details of your patient's lives: what's most important to them, what their other priorities are, what motivates them, what their financial situation is, and so on. Each patient is the expert in his or her own life.
Finally, we know from several studies that when patients are encouraged to be more involved and when their physicians are less prescriptive, patients do have better outcomes. We also know that this approach does not take any more time but, in fact, can be more efficient because the health care team is addressing the patient's agenda first and the patient's agenda is, after all, the real reason for the visit.1
In addition to being a more effective approach, it can also be a relief. As a physician, you may feel less frustrated because it is no longer your responsibility to make change happen. It is a joint process. When you create that partnership and get out of the role of simply telling patients what to do, you pave the way for the patient to make significant, lasting change.
Save 10% off your total NEW APF Merchandise
Wednesday - December 13, 2017 @ 07:00:29
We are excited to share with you the new APF Winter Items for sale.
Our hope is that you all will purchase and wear APF merchandise to help raise awareness about Porphyria.
Please note that all orders will go through APF employee Amy Chapman.Amy.Apf@gmail.com
This Week only save 10% off your total purchase!
Please follow the details below to complete your order. No items printed on the back of items due to hood covering.
****We DO NOT accept orders through Facebook****
Please submit your order and payment to Amy.APF@gmail.com or call 1-616-213-0030directly and leave a detailed message.
Please send an email and include the following: Full Name, complete mailing address, phone number.
For privacy purposes, please send in the email either the Visa/MC Number, Expiry Date, & CVV code on the back of your card.
If you prefer I can contact you by phone. NO CC info is EVER kept on file!
Order: Please send your items that you would like to order by quantity & size for each T-shirt.
**International orders may be priced higher allow 48 hours**
All orders include shipping prices for USPS only.
Long Sleeve Shirts~ are 6 ounces each 100% Cotton.
Colors are Medium Gray/Grey or Purple
Sizes available Adults Only- S, M, LG, X-LG, 2XL, 3XL- (X-LG. 2XL, 3XL are 3.00 each more)
Price Each- S, M, L 20.00$ Each ** X-LG. 2XL, 3XL are 3.00 each more**
Long Sleeve 100% Cotton Thick Zip Up Hoodie Colors are Medium Grey/Gray or Royal Purple.
Sizes available Adults Only- S, M , LG, X-LG, 2XL, 3XL- (X-LG. 2XL, 3XL are 5.00 each more)
Price Each- S, M, L 45.00$ Each ** X-LG. 2XL, 3XL are 3.00 each more**
Long Sleeve 100% Cotton Pull Over Hoodie Colors are Medium Grey/Gray or Royal Purple
Sizes available Adults Only- S, M, LG, X-LG, 2XL, 3XL- (X-LG. 2XL, 3XL are 5.00 each more)
Price Each- S, M, L 40.00$ Each ** X-LG. 2XL, 3XL are 3.00 each more**
Grey/Gray Soft Baseball cloth hats adjustable hat backside- 22.00$ Each (Fits Adults Sizes)
Wristbands- 2/6.00 or 3.00$ each Colors are Blue or Purple
Porphyria Car or Fridge Magnet 8.00$ each
ANOTHER WAY TO SUPPORT THE APF - AMAZONSMILE
Still doing your Christmas shopping? Many of you purchase numerous items from Amazon, not just books. Now you can support the APF through the AmazonSmile program! Amazon will donate 0.5-0.8% of the price of your eligible purchases to the APF, at no cost to you. Please make the APF your choice of a charitable organization. Support porphyria research while shopping!
Note, this program to provide donations to the APF will ONLY be available to shoppers who visit Amazon via a special web address, namely, www.smile.amazon.com instead of the normal www.amazon.com homepage.
It is easy and free! AmazonSmileis the same Amazon you know - same products, same prices, same service.
NORD gratefully acknowledges Dr. Michael Badminton, MBChB, PhD, FRCPath, Honorary Consultant and Clinical Lead, National Acute Porphyria Service (Cardiff), Medical Biochemistry & Immunology, University Hospital of Wales, for assistance in the preparation of this report.
Synonyms of Acute Intermittent Porphyria
AIP
Swedish Porphyria
General Discussion
Summary
Acute intermittent porphyria (AIP) is a rare metabolic disorder that is characterized by deficiency of the enzyme as hydroxymethylbilane synthase (also known as porphobilinogen deaminase). This enzyme deficiency can result in the accumulation of porphyrin precursors in the body. This enzyme deficiency is caused by a mutation in the HMBS gene which is inherited as an autosomal dominant trait (only one HMBS gene copy is affected). However, the deficiency by itself is not sufficient to produce symptoms of the disease and most individuals with a HMBS gene mutation do not develop symptoms of AIP. Additional factors such as endocrine factors (e.g. hormonal changes), the use of certain drugs, excess alcohol consumption, infections, and fasting or dietary changes are required to trigger the appearance of symptoms. Symptoms include severe abdominal pain, constipation, muscle weakness, a rapid heartbeat (tachycardia), behavioral changes, seizures, and damage of the nerves to muscles outside of the central nervous system (peripheral neuropathy). Treatment is focused on preventing attacks by educating patients to avoid potential triggers. Acute attacks usually require hospital care and can be effectively treated with intravenous hematin.
Introduction
AIP belongs to a group of disorders known as the porphyrias. This group of disorders is characterized by abnormally high levels of porphyrins and porphyrin precursors due to deficiency of certain enzymes essential to the creation (synthesis) of heme, a part of hemoglobin and other hemoproteins found in all cells. There are eight enzymes in the pathway for making heme and at least seven major forms of porphyria. The symptoms associated with the various forms of porphyria differ. It is important to note that people who have one type of porphyria do not develop any of the other types. Porphyrias are generally classified into two groups: the "hepatic" and "erythropoietic" types. Porphyrins and porphyrin precursors and related substances originate in excess amounts predominantly from the liver in the hepatic types and mostly from the bone marrow in the erythropoietic types. Porphyrias with skin manifestations are often referred to as "cutaneous porphyrias." The term "acute porphyria" is used to describe porphyrias that can be associated with sudden attacks of pain and other neurological symptoms. Two porphyrias can have cutaneous and acute symptoms, sometimes together. Most forms of porphyria are genetic inborn errors of metabolism. AIP is an acute, hepatic form of porphyria.
Signs & Symptoms
AIP can be associated with a range of symptoms and physical findings that can potentially involve multiple organ systems of the body. The course and severity of attacks is highly variable from one person to another. In some cases, particularly those without proper diagnosis and treatment, the disorder can potentially cause life-threatening complications. It is important to note the highly variable nature of AIP and that affected individuals may not have all of the symptoms discussed below. Affected individuals and parents of affected children should talk to their physician and medical team about their specific case, associated symptoms and overall prognosis.
The symptoms of AIP usually occur as episodes or attacks that develop over course of several hours or a few days. Affected individuals usually recover from an attack within days. However, if an acute attack is not diagnosed and treated promptly recovery can take much longer, even weeks or months. Most affected individuals do not exhibit any symptoms in between episodes. Onset of attacks usually occurs in the 20s or 30s, but may occur at or just after puberty. Onset before puberty is extremely rare. Attacks are much more common in women than men, probably because of the menstrual cycle hormones. Approximately 3%-5% of affected individuals, predominately women, experience recurrent attacks, which are defined as more than 4 per year, for a period of many years.
Abdominal pain, which is usually severe, is the most common symptom associated with AIP and often the initial sign of an attack. Abdominal pain is usually severe, steady (unremitting) and widespread (diffuse). Less often, abdominal pain is described as cramping. Pain may also occur in the neck, lower back, buttocks, or arms and legs.
Gastrointestinal symptoms are also common during an attack and can include nausea, vomiting, constipation or diarrhea, and abdominal swelling (distention). A painful blockage or obstruction (ileus) of part of the small intestines may also occur. Difficulty passing urine (urinary retention) can also occur.
Neurological symptoms may also develop including damage to the nerves outside the central nervous system (peripheral neuropathy). Peripheral neuropathy is characterized by numbness or tingling and burning sensations that usually begin in the feet and sometimes the arms. Affected individuals may develop muscle weakness in the legs that may progress to affect the arms and the trunk of the body, eventually causing partial loss or impairment of motor function (motor paralysis). In rare cases, the muscles used to breathe can become involved and potentially cause life-threatening respiratory failure which requires mechanical ventilation.
Some individuals develop psychological symptoms including irritability, depression, anxiety, insomnia, hallucinations, paranoia, disorientation, and altered consciousness ranging from excessive drowsiness (somnolence) to agitation or, in severe cases, coma.
Affected individuals may also experience a faster than normal heart rate (tachycardia) and irregular heartbeats (cardiac arrhythmias). Seizures have also been reported. Abnormally low sodium levels (hyponatremia) may develop rapidly during an attack and contribute to the onset of seizures.
Individuals with chronic AIP may develop complications that occur after many years (long-term complications) such as high blood pressure (hypertension), kidney damage potentially resulting in kidney failure, and a form of liver cancer known as hepatocellular carcinoma (HCC).
Causes
AIP is a multifactorial disorder, which means that several different factors such as genetic and environmental factors occurring in combination are necessary for developing symptoms of the disorder. Individuals with AIP have a mutation in the HMBSgene. Genes provide instructions for creating proteins that play a critical role in many functions of the body. When a mutation of a gene occurs, the protein product may be faulty, inefficient, or absent. Depending upon the functions of the particular protein, this can affect many organ systems of the body.
However, the majority of people with a mutation in this gene do not develop symptoms of AIP; additional factors, often called triggers are also required to cause symptomatic acute porphyria. These factors are not necessarily the same for each individual, and susceptibility to specific triggers may vary during a patients lifetime. Most of these triggers are believed to stimulate increased heme production (synthesis) in the liver and include certain drugs, excessive alcohol consumption, fasting or dietary (e.g. caloric restriction), stress, infections or certain hormonal (endocrine) factors.
The HMBS gene mutation that predisposes individuals to developing AIP is inherited as an autosomal dominant trait. Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary for the appearance of the disease. The abnormal gene can be inherited from either parent, or can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy regardless of the sex of the resulting child.
The HMBS gene creates (encodes) the enzyme porphobilinogen deaminase (PBG-D), which is also known as hydroxymethylbilane synthase or uroporphyrinogen I synthase. This enzyme is the third enzyme is the process of heme biosynthesis. Mutations in the HMBS gene lead to deficient levels of PBG-D in the body, which in turn can lead to the accumulation and release of porphyrin precursors, 5-aminolevulinic acid (ALA) and porphobilinogen (PBG) from the liver.
The elevation of porphyrin precursors alone is not sufficient for the development of symptoms of AIP. As discussed above, triggering factors are required for symptom development. The exact, underlying reasons why symptoms develop in some individuals with AIP are not fully understood. There are several theories as to the underlying pathogenesis of AIP. One theory states that a specific porphyrin precursor (most likely ALA) is a hepatic neurotoxin that damages nerve tissue. This theory is supported by the information obtained from patients who have had liver transplant, which corrects both the clinical and biochemical features of the condition. A second theory suggests that heme deficiency in nerve cells (neurons) contributes to the development of symptoms. More research is necessary to determine the exact underlying mechanisms that are involved in the development of symptomatic episodes in individuals with AIP.
Affected Populations
The exact incidence and prevalence of symptomatic AIP is unknown. In Europe the prevalence is estimated to be approximately 5.9 per million people in the general population. Prevalence is greatest in Sweden due to a founder effect. AIP can occur in individuals of all racial and ethnic backgrounds, although it is rarely reported in African-American individuals. Women are affected by symptomatic AIP more often than men. The disorder is most common in young or middle-aged women.
Related Disorders
Symptoms of the following disorders can be similar to those of AIP. Comparisons may be useful for a differential diagnosis.
The acute attacks that characterize AIP are similar to those seen in three other forms of porphyria specifically variegate porphyria, hereditary coproporphyria, and ALA-Dehydratase deficiency porphyria. Collectively, these four forms of the porphyria are classified as the acute porphyrias. (For more information on these disorders, choose the specific disorder as your search term in the Rare Disease Database.)
Tyrosinemia type I is a rare autosomal recessive genetic metabolic disorder characterized by lack of the enzyme fumarylacetoacetate hydrolase (FAH), which is needed for the final break down of the amino acid tyrosine. Failure to properly break down tyrosine leads to abnormal accumulation of tyrosine and its metabolites in the liver, including a heme precursor ALA, potentially resulting in severe liver disease. Tyrosine may also accumulate in the kidneys and central nervous system. Symptoms and physical findings associated with tyrosinemia type I appear in the first months of life and include failure to gain weight and grow at the expected rate (failure to thrive), fever, diarrhea, vomiting, an abnormally enlarged liver (hepatomegaly), and yellowing of the skin and the whites of the eyes (jaundice). Tyrosinemia type I may progress to more serious complications such as severe liver disease, cirrhosis, and hepatocellular carcinoma if left untreated. Untreated children can also suffer neurological crises similar to those seen in acute porphyria. Treatment with nitisinone and a low-tyrosine diet should begin as soon as possible after the diagnosis is confirmed. (For more information on this disorder, choose tyrosinemia as your search term in the Rare Disease Database.)
Lead toxicity can cause symptoms that mimic acute porphyria (acute abdominal pain, constipation, neuropathy). Lead inhibits several of the enzymes of haem biosynthesis, which can therefore result in an increase in urine coproporphyrin and 5-aminolevulinic acid excretion, but not porphobilinogen excretion. It can also cause an increase in erythrocyte protoporphyrin concentration, although this is all the zinc-chelate form (zinc-protoporphyrin). The definitive test for lead poisoning is blood lead measurement.
Diagnosis
A diagnosis of AIP can be difficult because most symptoms are nonspecific and occur episodically. A diagnosis is usually based upon identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation and certain specialized tests. AIP should be suspected in individuals with unexplained abdominal pain, especially when occurring along with muscle weakness, psychological symptoms, neurological findings or unexplained hyponatraemia. Dark or reddish urine in such individuals is also suggestive of AIP. However absence of this feature does not exclude AIP.
Clinical Testing and Workup
Screening tests to measure the levels of the porphyrin precursor porphobilinogen (PBG) in urine are essential to confirm a diagnosis of acute porphyria. Acute attacks are always accompanied by increased excretion of PBG in AIP. If urinary PBG excretion is increased, then further testing (fecal and blood porphyrin measurement) is necessary to distinguish AIP from variegate porphyria or hereditary coproporphyria. Delta-aminolevulinic acid (ALA) excretion will also be elevated in urine samples from individuals with AIP, but measurement is less widely available and is not essential. These tests can be performed on a random (spot) urine sample that should be protected from light after collection and during transport to the laboratory. There is now good evidence that once urine PBG excretion is increased in AIP it takes many years to return to normal. Increased urine PBG excretion in a known AIP patient is therefore not necessarily diagnostic of an acute attack.
Family testing
Molecular genetic testing is not essential to confirm a diagnosis as the porphyrin biochemical findings are characteristic. However molecular genetic testing to detect a mutation in the HMBS gene is usually required so that family members can be offered testing for this mutation. Genetic testing is available mainly from laboratories specializing in porphyria diagnosis.
Patients and family members who have inherited AIP should be counseled on how to limit their risk of any future acute attacks. This should include information about AIP and what causes attacks, how to check if a prescribed medication is safe or unsafe and details of relevant patient support groups.
Standard Therapies
Treatment
The treatment of AIP is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, neurologists, hematologists, hepatologists, psychiatrists, and other healthcare professionals may need to systematically and comprehensively plan an affected patients treatment. Genetic counseling may benefit affected individuals and their families.
The objective of treatment is to manage symptoms, prevent complications and to suppress heme creation (synthesis) in the liver with hematin, which reduces the production of porphyrin precursors. Initial treatment steps also include stopping any medications that can potentially worsen AIP or cause an attack and ensuring proper caloric intake, which can include intravenous infusion of sufficient nutrients (glucose and salt). Carbohydrate loading in conjunction with good pain medication may be sufficient for mild attacks.
An acute neurovisceral attack often necessitates hospitalization and may require treatment with hematin. In the United States, affected individuals may be treated with Panhematin (hemin for injection), an enzyme inhibitor derived from red blood cells that is potent in suppressing acute attacks of porphyria. Panhematin almost always returns porphyrin and porphyrin precursor levels to normal values. The U.S. Food and Drug Administration (FDA) originally approved panhematin for the treatment of recurrent attacks of AIP in related to the menstrual cycle in susceptible women. Because of its potency, it is usually given after a trial of glucose therapy and should be administered only by physicians experienced in the management of porphyrias in a hospital setting. Panhematin is manufactured by Lundbeck, Inc.
Heme arginate (Normosang, Orphan Europe) is another heme preparation that can be used to treat individuals with AIP. Heme arginate is not available in the United States, but is often used in other countries where Panhematin is not available.
Treatment for AIP also includes drugs to treat specific symptoms such as certain pain medications (analgesics), anti-anxiety drugs, anti-hypertensive drugs, and drugs to treat nausea and vomiting, tachycardia, or restlessness. Medications to treat any infections that may occur at the same time as an attack (intercurrent infection) may also be necessary. Although many types of drugs are believed to be safe in individuals with AIP, recommendations about drugs for treating AIP are based upon experience and clinical study. Since many commonly used drugs have not been tested for their effects on porphyria, they should be avoided if at all possible. If a question of drug safety arises, a physician or medical center specializing in porphyria should be contacted. A list of these institutions may be obtained from the American Porphyria Foundation (see the Resources section of this report). The Foundation also maintains an Acute Porphyria Drug Database.
Additional treatment for individuals undergoing an attack including monitoring fluid and electrolyte balances. For example, if individuals develop hyponatremia, which can induce seizures, they should be treated by saline infusion.
In some cases, an attack is precipitated by a low intake of carbohydrates in an attempt to lose weight. Consequently, dietary counseling is very important. Affected individuals who are prone to attacks should eat a normal balanced diet and should not greatly restrict their intake of carbohydrates or calories, even for short periods of time. If weight loss is desired, it is advisable to contact a physician and dietician. Premenstrual attacks often resolve quickly with the onset of menstruation. Hormone manipulation may be effective in preventing such attacks and some affected women have been treated with gonadotropin-releasing hormone analogues to suppress ovulation and prevent frequent cyclic attacks. Some individuals who experience recurrent attacks may benefit from regular hematin infusion. This is sometimes recommended for women with severe symptoms during the time of their menses.
If a proper diagnosis has not been made, AIP can be particularly dangerous, especially if drugs which aggravate the disorder are administered. The prognosis of AIP is usually good if the disorder is recognized before severe nerve damage has occurred and if treatment and preventive measures are begun. Although symptoms usually resolve after an attack, some individuals may develop chronic pain. Nerve damage and associated muscle weakness from a severe attack improves over time, but such improvement may take many months to resolve fully.
Liver transplantation has been used to treat some individuals with AIP, specifically individuals with severe disease who have failed to respond to other treatment options. A liver transplant in individuals with AIP is an option of last resort. Affected individuals who experience kidney failure may require a kidney transplant. Some individuals have required a combined kidney/liver transplant.
Investigational Therapies
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
For information about clinical trials sponsored by private sources, in the main, contact: www.centerwatch.com
For more information about clinical trials conducted in Europe, contact: https://www.clinicaltrialsregister.eu/
NEW! 2017 APF Winter Merchandise
Monday - December 11, 2017 @ 15:10:22
APF Merchandise for Sale!
Our hope is that you all will buy and wear APF merchandise to help promote and raise awareness about Porphyria. Please note that all orders will go through APF employee Amy Chapman. Amy.Apf@gmail.com
****We DO NOT accept orders through FB****
Please submit your order and payment to Amy.APF@gmail.com or call 1-616-213-0300 directly and leave a detailed message.
Please send an email and include the following: Full Name, complete mailing address, phone number.
Order: Please send your items that you would like to order by quantity & size for each T-shirt. For privacy purposes, please send in the email either the Visa/MC Number, Expiry Date, & CVV code on the back of your card. If you prefer I can contact you by phone.NO CC info is EVER kept on file!
**International orders may be priced higher allow 48 hours**
All orders include shipping prices for USPS only
Long Sleeve Shirts~ are 6 ounces each 100% Cotton. Colors are Medium Gray or Purple
Price Each- S, M, L 20.00$ Each ** X-LG. 2XL, 3XL are 3.00 each more**
Long Sleeve 100% Cotton Thick Zip Up Hoodie Colors are Medium Gray or Royal Purple.
Price Each- S, M, L 45.00$ Each ** X-LG. 2XL, 3XL are 5.00 each more**
Long Sleeve 100% Cotton Pull Over Hoodie Colors are Medium Grey/Gray or Royal Purple
Price Each- S, M, L 40.00$ Each ** X-LG. 2XL, 3XL are 5.00 each more**
Greg/Gray Soft Baseball cloth hats adjustable hat backside- 22.00$ Each
(Fits Adults Sizes)
Wristbands- 2/6.00 or 3.00$ each Colors are Blue or Purple
Porphyria Car or Fridge Magnet 8.00$ each
Get yours today for a loved One!
**UPDATED** Patient Day January 13, 2017 Orlando FL PLEASE READ!
Friday - December 8, 2017 @ 06:00:47
Saturday January 13, 2018
Patient Day
10:00 am
Welcome D. Lyon-Howe, American Porphyria Foundation
10:05 am
American Porphyria Foundation Overview
Session 1- Heme Biosynthesis & the Erythropoietic Porphyrias
Co-Chairs: E. Minder, MD & S. Keel, MD
10:15 am
Heme Biosynthesis and the Porphyrias 101J.D. Phillips, PhD
10:45 am
Genetics 101H. Naik, MS, CGC
11:00 am
Erythropoietic Porphyrias Overview M. Balwani, MD, MS
11:30 am
EPP Research J. Marcero
11:45 am
Effective & Emerging Therapies for the Treatment of Erythropoietic Porphyrias E. Minder, MD, MS
12:15 pm
Q & A moderated by Co-Chairs
12:45 pm
Lunch
Session 2- the Acute Hepatic Porphyrias
Co-Chairs: D.M. Bissell, MD & C. Levy, MD
1:45 pm
Diagnosis of the Porphyrias M. Badminton, MBChB, PhD
Research Overview and Patient PerspectiveTBD and Sharon Dill, Patient Representative
3:45 pm
Q & A moderated by Co-Chairs
4:30 pm
Closing remarksAmerican Porphyria Foundation
APF just got a makeover!
Thursday - December 7, 2017 @ 07:00:50
FRESH NEW LOOK
Same dedication to the Porphyria community!
The American Porphyria Foundation is launching a brand new look today. You will see this refresh across our communications including the website, seven Facebook groups, Porphyria Post, Twitter, Purple Light Blog, Google and YouTube. We are proud of our new look that represents the people we serve and the group of rare diseases we represent. We remain steadfast in our dedication to support individuals and families whoare impacted by Porphyria through physician and patient education, awareness campaigns, strong advocacy and a relentless focus on research.
Stay tuned for more exciting updates from your APF team in 2018! As always, thank you for your wonderful support.
2018 Heme Biosynthesis and the Porphyrias: Recent Advances - ORLANDO, FL
Mark your calendars to join the American Porphyria Foundation and the Genetic Disease Foundation along with expert physicians from around the world! On Saturday, January 13, 2018 there will be a Clinical Day for Patients which will include in-depth discussion on Erythropoietic and Acute Hepatic Porphyrias with domestic and international experts, interactive Q&A Sessions and much more. Registration is COMPLIMENTARY. If you plan on attending the clinical day for patients, please contact Edrin at the APF office today to register.
"Remember.Research is the key to your cure!"
Jasmin Barman EPP
Tuesday - December 5, 2017 @ 09:53:20
After having had the pleasure of getting to know many of you in person at the FDA workshop for EPP-patients last October in Maryland, I am glad to be given the opportunity to share my personal story as an EPP patient, porphyria researcher and patient advocate with you.
Like most EPP patients, it took me very long between the first phototoxic reaction at the age of 2.5 years and my diagnosis. One night in spring 2006, in my final year as a molecular biology student in Heidelberg (Germany), yet again I could not sleep because of the intense pain caused by the strong sunlight I was exposed to during my way home from university, despite having worn thick, long clothing, using an umbrella and walking in the shadow whenever possible. Like so many times before, I got up and searched the Internet for an explanation to my strange sun allergy. Excitingly, this time, I found a new article on Wikipedia, with the unfamiliar title erythropoietic protoporphyria. Those eight sentences written in lay terms just two weeks earlier by the beloved founder of the German EPP patient organisation, Verena Schmeder, described my life and changed it in a way I would never have guessed: Today, I am in charge of the laboratory conducting all porphyria diagnoses in Switzerland, act as scientific advisor for the Swiss Society for Porphyria, am a co-founder of the International Porphyria Patient Network and represented the patient perspective during the approval process of afamelanotide (Scenesse) at the European Medicines Agency (EMA).
But first things first: After finding out about EPP, it was overwhelming to meet other sufferers for the first time in my life. It was and still is impressive for me to see, how similar the experiences and coping strategies of EPP patients are: Since the majority of people around us do not believe how painful EPP is and how much it restricts all aspects of daily life, we tend to hide the condition and try to find our own ways in this world poisoned by light. Being a scientist, of course I also was fascinated to learn about all the aspects already known about EPP, like the fact that visible light is causing the phototoxic reactions and not UV radiation: Finally an explanation of why sunscreens never helped! And of course, I was intrigued by the many things not yet understood about EPP. In October 2006, Italian porphyria patients organized the first international porphyria patient day in Rome. There, I had the chance to discuss a few research ideas with the porphyria expert Prof. Elisabeth Minder from Switzerland. You can imagine my excitement, when she offered me a PhD position in her lab in Zurich! I joined her team in 2007 and my main area of research became iron metabolism and gene regulation in EPP.
During my time as a student in Elisabeth`s group, she conducted the first clinical trial ever testing afamelanotide (Scenesse) in EPP. As an employer of the institute, I did not participate in the trials. However, I met fellow patients and was intrigued by the dramatic benefit this new therapeutic approach seemed to have on them. Then, thanks to enormous efforts by Elisabeth, nearly all patients in Switzerland could enrol in an early access program in 2012 and receive the afamelanotide treatment. Of course, I jumped at the opportunity but was rather sceptical and had limited expectations about the effects in the beginning but after overcoming the fear to be exposed to sunlight I experienced the same amazing, life-changing effects described by the others: Instead of a few minutes until the first symptoms started, I could now spend hours with friends and family outdoors! I also almost lost my aversion to be exposed to light because even if I developed a reaction, it would be much less painful and only last until the next morning and not up to two weeks as without the treatment. In the beginning, it felt crazy to choose the side of the street I wanted to walk on and not being restricted to the shadowy side! Thanks to the treatment I now have an almost normal life, I am able to make appointments to visit partner laboratories during day time, teach at the University of Zurich in spring and even attend scientific conferences in the summer, like the Porphyrins & Porphyrias congress this past June in Bordeaux.
In 2012, the approval process for afamelanotide started in Europe: Between 2006 and 2012, five clinical trials in Europe and the US with more than 350 participants were concluded, and all showed that afamelanotide helps EPP sufferers to stay outdoors for longer and to experience less pain. In contrast to the FDA, the EMA does not have a standard policy to invite rare disease patients when evaluating a potential therapy for their condition. In 2013, Dr. Rocco Falchetto, president of the Swiss Society for Porphyria, and I organized an international patient day in Switzerland. All representatives of the attending porphyria patient organisations, amongst others Desiree Lyon from the APF, took the opportunity and together we wrote a letter to EMA, asking them to include patients in the process. Indeed, in 2014, EMA for the first time in their history invited another EPP patient and me to participate at a full regulatory committee meeting directly prior to the drug approval decision. We think that bringing in the patient perspective was crucial to make regulators understand the true burden of EPP and the real benefit that the treatment with afamelanotide brings! Frustratingly however, the excessive and complex post-marketing obligations demanded as a condition for approval by EMA make the treatment nearly inaccessible for European sufferers - three years after approval, only patients from the Netherlands have unrestricted access. Currently, EPP patients all over Europe organize protests, engage in political discussions and start media campaigns to make their voice heard. Our experience also shows: In every country, dedicated physicians are absolutely essential to the process and, if they really want to, they find ways to provide the treatment to their patients! The best examples are Italy, where patients thanks to Prof. Gianfranco Biolcatis personal engagement had access from 2009 on, even before the approval by EMA, and the early access program Switzerland leveraged by Prof. Minder, which is not even an EU member state.
For me, it is inacceptable to not alleviate rare disease patients from their suffering if a possibility exists to treat their condition. What is the rational for research if patients do not benefit from it when a drug is successfully developed? Patients potentially risk their health in drug trials, agreeing to test new substances and hoping to contribute to a better future for themselves and their fellow sufferers, in particular children, who are affected the most. In the current health care systems however, patients have to wait for very long and even might never gain access to a safe and effective treatment, if interests other than the patients health are prioritized. Again and again we have to explain and justify ourselves to so many representatives of authorities, regulators, committee members and staff of health insurance companies who are in the position to either give us a life or send us back into pain and darkness. No porphyria patient chooses the condition he or she suffers from. Therefore, whenever possible, I support the efforts of EPP patients and other advocates with my knowledge and experience. Seeing which difference the therapy makes for the sufferers motivates me again and again to give my best every day. And I am optimistic that with the European experience and the exchange the patient organisations are now having between each other, approval and access will be faster and easier to achieve for other countries.
This is how I first became a scientist, then a patient, and now a porphyria researcher and patient advocate. Because of EPP, I will never be the outdoor biologist catching new species in the mud of a tropical rainforest that I always wanted to become. But because of the condition I got to know so many great people in the patient and research community, found a fascinating research area and witnessed the development of a new drug! I am looking forward to the many exciting new developments in the field of porphyrias and hope to meet many of you in January in Orlando!
It's very difficult for patients to do what they don't understand, so the first step in equipping patients to take on a more active role in their health care is to educate them. Start by communicating to patients that education is perhaps as important to their health as getting their prescriptions filled. They need to know all they can about their disease.
But just as patient-centered care can be more effective, patient-centered education is better education. The old education program, where you bring people in, sit them down and lecture to them, doesn't work any better than bringing them in, sitting them down and telling them to lose 20 pounds. Instead, the patient's needs should drive the education. For example, our center is testing diabetes education courses based entirely on questions from the audience. We do have a checklist of topics we want to cover, but we address those topics in the context of patient questions rather than through an impersonal lecture. Patients aren't interested in their disease from an intellectual perspective, as we are. They want to know about themselves. What does this mean to me? How's this different for me? How's it going to affect my life?
Four of the most important lessons patients with chronic diseases need to understand are the following:
1.Their illness is serious. There are still patients out there who believe they have the not-so-serious kind of diabetes. If they don't believe it is a problem, they will never make changes to improve their health.
2.Their condition is essentially self-managed. Every decision patients make throughout the day, from what they eat to whether they walk or ride the bus, has an influence on their health. Communicate to patients that they are the most important individuals in managing their illnesses.
3.They have options. There is rarely one perfect way to treat a condition. In the case of diabetes, for example, patients can be treated through diet and exercise, oral medication, insulin and so on. Patients need to understand the different treatment options available and should be encouraged to look at the personal costs and benefits of each. Only the patient can decide if the benefits are greater than the costs.
4.They can change their behavior. Rarely do patients leave the doctor's office and immediately enact whatever change was recommended. The reality is that it often has to be spread out into a series of steps. Teach patients that significant behavioral changes can be made by setting goals, taking that first step and figuring out what you learn about yourself along the way.
Empowering patients with information
One way to help patients focus and begin thinking about their health care goals is to talk with them about their individual health measures (e.g., blood pressure, LDL, HbA1c) and what those numbers mean. At our center, for example, we give patients a handout that lists the critical measures for their condition (ideal and actual), explains what those numbers mean and offers strategies for improvement. When faced with this information, patients can see for themselves where they are struggling and what they can do to better their scores.
A sample page from our handout is shown here.
Blood Pressure
Actual: _________________ mm/Hg
Ideal: 130/85 or lower
My goal is: ______________
A blood pressure reading has two numbers. The top number is called systolic blood pressure. This is the amount of pressure against the blood vessel walls when your heart pumps. The bottom number is called diastolic blood pressure. This is the amount of pressure against the blood vessel walls when your heart relaxes, that is, between heart beats.
In general, high blood pressure means that systolic blood pressure, diastolic blood pressure or both may be too high. For people with diabetes, high blood pressure is 130/85 or higher. High blood pressure increases your risk for strokes, heart attacks, kidney damage and eye disease.
To lower your blood pressure you can:
·Eat less salt,
·Take blood pressure medicine,
·Exercise,
·Stop smoking,
·Monitor blood pressure,
·Drink less alcohol,
·Maintain reasonable weight,
·Other: _________________________________
Helping patients set goals
In the patient-centered model of care, the driving force behind each patient visit is the patient's agenda or goals related to his or her condition. Ideally, the goal is clearly displayed in the patient's chart, and each person who handles the chart plays a part in supporting the patient in that goal, asking, How did it go? What have you done this week? How can we help you do better?
You might be thinking, My patients don't have goals, but they do. Even noncom-pliant patients have goals. Probably the best definition of noncompliance is a doctor and patient working toward different goals.
The process of setting self-management goals with the patient involves essentially two steps.
1.Start at the problem. Rather than beginning the patient encounter focused on lab values or weight or blood pressure readings, begin by saying, Tell me what concerns you most. Tell me what is hardest for you. Tell me what you're most distressed about and what you'd most like to change. You'll get to the lab values and other issues later, but it will be in the context of the patient's personal goal, which will make it more meaningful for the patient.
As you begin to get a sense of the patient's concerns, explore those issues together. Ask, Is there an underlying problem? Do you really want this problem to be solved? What's the realissue?
2.Develop a collaborative goal. Once you have worked with the patient to identify the real problem, your instinct may be to try to solve it, but don't. Don't try to fix it. Don't just say, It will be OK. Instead, validate the patient's feelings and his or her capacity to deal with the problem, and continue asking questions that will lead the patient to his or her own solution. Ask, What do you think would work? What have you tried in the past? What would you like to try?
It's always more meaningful when patients find the ah ha! on their own, so give them that chance. Encourage them to come up with ideas first, then offer your own suggestions or additional information that they may need. You can say this works for some people or have you tried this? or here's why I don't think that's a good idea. The important thing is to give the patient the opportunity to say no and to make the final decision on what goal to try.
Ultimately, at the end of the conversation, the patient should be able to tell you one step he or she is going to take. It should be very specific. If the patient says, I'm going to exercise more, ask what that means. Will they exercise four times a week? What activity will they be doing? How far will they walk? Help them to come up with a specific plan that they have created for themselves. It may not be the ultimate goal you would have chosen for the patient, but it's one they are more likely to accomplish. At the next visit, then, you can build on that.
Who actually works with patients to set their goals, whether you or the nurse or the diabetes educator, is perhaps less important than the fact that patients are encouraged to be more involved. The emphasis on self-management goals suggests that the visit is for them. It is their agenda, and they are active participants in the outcome.
Series overview
This article is part of an FPM series that followed Family Care Network, a northwest Washington state group without walls, as it tackled a 13-month quality improvement project focused on chronic disease care. The project was headed by the Institute for Healthcare Improvement and involved approximately 30 organizations nationwide.
The patient-centered model of care, and the emphasis on empowering patients to find their own solutions, may go against your instincts as a physician. As health care professionals, we often feel most helpful when we've given advice. The truth is, however, that we don't really help people solve their problems or make lasting changes in their lives by telling them what they should do. Ultimately, patients need to find their own solutions and motivation and must take responsibility for their health. We must empower them to do just that.
Martha Funnell, a certified diabetes educator, is the director for administration at the Michigan Diabetes Research and Training Center, University of Michigan Health System at Ann Arbor. The MDRTC has been funded by the National Institutes of Health since 1977 and is one of six such centers in the United States.
REFERENCE
1. Wagner EH, Austin BT, Von Koroff M. Improving outcomes in chronic illness. Managed Care Quarterly.1996;4(2):1225.
The new drug, Givosarin, is being researched to prevent acute porphyria attacks
Wednesday - December 6, 2017 @ 17:56:23
Very Important!!! Be A Medical Hero!!!
Research sites are opening for the Givosarin drug to prevent attacks in Miami, Wake Forest , NC, Galveston, TX, New York, Philadelphia, San Francisco, Ann Arbor, MI, Salt Lake City, Little Rock, Boston,
The new drug, Givosarin, is being researched to prevent acute porphyria attacks and hopefully the chronic pain associated with acute porphyria..aip, hcp, vp, Adp.
So far, the results of a trial have been very impressive. We cannot have new treatment without You as a volunteer. Participation and travel is free .
Call the APF toll free 866-APF-3635.
INTRODUCING SHADOW JUMPERS!
Tuesday - November 28, 2017 @ 06:00:11
INTRODUCING SHADOW JUMPERS!
Hey everyone! The APF is creating our own version of a "Kid's Corner" focused on children with EPP. Our website will soon have a section called Shadow Jumpers. This will be a place for kids to learn from others with EPP, to share their own tips and tricks on things like what to wear, what to do for fun, insight for parents and so much more. We need your help to get started!
We are looking for YOUR tips and tricks on managing EPP. If you are a caregiver, please ask your child with EPP to share their ideas through you. These could include
What clothes and trends do you use when covering up from the sun?
·What are the ways you adapt to do certain activities outside?
·What are fun things you do to pass the time inside while the sun is out?
·What have been your best vacations?
·How do you manage in the car? Learning to drive?
·What do you do when you feel sad or left out?
·How do you manage going to school?
·How do you describe your EPP to other people?
·and anything else you think may be helpful to others!
We encourage you to send pictures or videos pertaining to your tips!
We can't wait to hear from you!
Stay tuned for more from Shadow Jumpers!
#GivingTuesday
Monday - November 27, 2017 @ 07:00:13
The APF is participating in #GivingTuesday on Tuesday, November 28 - a global giving movement that has been built by individuals, families, organizations, and communities in all 50 states and in countries around the world. Millions of people have come together to support and champion the cause they believe in helping. We hope that you choose the APF! Each and every donation to the APF will have a huge impact toward helping people impacted by Porphyria. Get ready to join the movement and share with your community!!
Part 6 Everyone, Regardless of Age, Can Be Invisibly Disabled
Friday - November 24, 2017 @ 07:00:21
6. Everyone, Regardless of Age, Can Be Invisibly Disabled
All of these things are extremely problematic because the chances are high that everyone living will have an invisible disability eventually. As with everything, the longer you live, the more chances you have to experience something.
However, just because age is correlated with illness doesnt mean that age alone causes illness.
Certain illnesses are just a matter of time and chance, yet health is treated as a static reality and youth as invulnerability. If you only view illness as an age-related event, you cant fully advocate or recognize the millions of children and young adults who are afflicted.
Even doctors who know I have chronic illnesses have still referred to me as healthy, as if my lack of years automatically cancels out the fact that some days Im about as mobile as a corpse.
Just because some of us are young doesnt automatically make us healthy.
It erases our experiences and makes us feel even more useless when compared to other people our age. We arent capable of the same things the rest of our age group is, and it hurts to be reminded of that.
These Problems Arise From the Illusion of Health
Health is assumed to be the default state, when in actuality, illness is much more likely and more common than realized.
As a chronically ill person, health is just a myth to me some fairy tale. I dont know how many others feel that way, but I bet its a lot.
Health, as commonly defined, is unattainable for those of us with painful chronic illnesses. Yet that doesnt mean all is lost. Become a better access ally.
Lessen the impact these problems have by realizing that chronic illnesses affect the entire being: heart, mind, body, and soul. They change the ways we can move in the world, so have consideration for those who have no visible signs of illness. Our lives depend on it.
Dont assume that were lying when we tell you we cant do something. Instead, propose an alternative that helps us. Ask what you can do around the house or offer to give us a massage.
Though we may be smiling and laughing, that doesnt mean were not in pain. It doesnt mean were healthy.
We are diverse as any other population, and we deserve information tailored to our needs and desires just as much as you do.
Keep us in mind, even though our struggles are invisible to you!
We hope you enjoyed reading over the six part series and know your never alone!
Part 5. The Absence of Invisible Illness in Accessibility Talks is Problematic
Thursday - November 23, 2017 @ 10:54:28
5. The Absence of Invisible Illness in Accessibility Talks is Problematic
Speaking of ineffective, even those who are active disability advocates fail to take into account the intersections of disability with other identities and realities.
Even certain types of disability are pushed to the margins. The most common invisible illnesses spoken of are mental disorders like schizophrenia and bipolar disorder, along with the neurodivergent experiences of autistic people.
But this means that anyone without a popularized chronic illness is marginalized within both media and disability movements.
There are plenty of disabled people saying they wanted to be treated like theyre normal, but for those of us with chronic pain, thats the opposite of a solution. Treating us like normal people, or even other disabled people, will absolutely make things worse.
Recently, I attended a disability festival. It was the most disability-inclusive environment Id ever been in, with sections for the deaf, those with low vision, those in wheelchairs, and more. Yet even they failed to take chronic pain sufferers into account: the chairs were rigid and hard. I nearly lay down on the floor to relieve the pain those chairs caused.
Until you can experience the pain of sitting, the pain of a breeze, you have no idea what chronic pain can do to a body.
Happy Thanksgiving 2017
Wednesday - November 22, 2017 @ 07:00:14
Happy Thanksgiving everyone 2017
Part 4. The Pain Scale Is Useless to Us
Tuesday - November 21, 2017 @ 16:14:09
4. The Pain Scale Is Useless to Us
Theres that pain scale, numbered one through ten, with increasingly dour expressions on the faces of the metaphorical patient. But the problem is that this scale is based on being healthy with a temporary illness. Its not properly designed for those who never have a day pass without being in pain.
Those of us with chronic illnesses are masters of coping. We learn to smile through the pain and anguish, we conserve what little energy we have by not bothering to cry, and we count out our spoons to figure out how much we can get done in a day.
Part of the reason its so hard to believe that many of us are truly sick is because of the ways we cope with our pain. Its more than most people will ever experience, yet we have no choice but to bear it.
We only break down when we have the spoons to break down. Even our doctors become lightweight suspicious or reluctant when we come across as cheerful even while our bodies feel like theyre on fire.
Theres this meme that went around showing an improved pain scale for people with fibromyalgia. The face was still smiling all the way up to ten. And thats the reality. If we make a sour face or actually cry, it means the pain has gone beyond the scale itself.
It also fails to account for all the different types of pain we experience. Theres the saying that the Inuit have many names for snow; well, I have a shitload of names for pain.
But until theres a more commonly understood, nuanced key for pain, there will be a disconnect between patients and doctors, loved ones and family. Medications or help that are sorely needed might be passed over because of a misunderstanding about how pain affects people differently.
You can provide better care to your loved ones if you can grok that just because theyre smiling doesnt necessarily mean they feel better.
Talks can become quite ineffective without proper language or tools. And right now, treatment based on pain scales is incredibly ineffective.
The American Porphyria Foundation will be closed on Wednesday, November 21, 2017 in observance of Thanksgiving and will re-open for normal business hours on Monday, November 27, 2017.
For emergency medical attention, please go to your nearest emergency room and contact your primary care physician. For all other issues, please direct your needs to the APF office staff via email. Your messages will be answered accordingly.
We hope that you enjoy this Thanksgiving holiday with your loved ones.
Please see below some additional announcements from the APF.
Want your physician to become better educated about your disease?
Personally invite them to this important symposium!!!
2018 Heme Biosynthesis and the Porphyrias: Recent Advances
Orlando, Florida - January 12-14, 2018
This three day symposium will focus on the most recent findings on the molecular biology and regulation of heme biosynthesis and the clinical features, pathophysiology, and current treatment of the acute hepatic and erythropoietic porphyrias. Day 1 will be devoted to the clinical aspects and current treatments of each Porphyria. Days 2 and 3 will focus on the latest research findings on heme biosynthesis and emerging molecular-based treatments for the porphyrias. Also, there will be a special session for patients on the latest advances in the Porphyrias.
This symposium is open to all healthcare professionals including clinicians, researchers, nurses, genetic counselors, Junior Faculty, Pre/Post Doctoral students, fellows and medical students.
The Symposium offers up to 25.5 CME Credits. The Registration fee includes meals and the poster reception. Orlando has an international airport and outstanding attractions such as Disney World and Universal Studios. The hotel rates are attractive ($133/night plus tax, single or double), with parking, Internet, and resort fees included. The room rate is available for three days after the symposium, on a first come basis.
To share this information with your doctor, you can provide them with the registration link or print and share the attached brochure and take to your appointments with you: www.porphyriassymposium.com
Members in the Atlanta, GA area are invited to attend a Patient Education and Support Meeting. There will be a presentation and question/answer session with anon-site Porphyria expert- Dr. John Phillips. Come meet old and new friends who share your experiences with Porphyria. Please RSVP to Edrin Williams in the APF office at 1.866.APF.3635 oredrinw@porphyriafoundation.org. We look forward to seeing you!
ALNYLAM TRIALS
The Alnylam Trials forGivosarinwill begin soon. The purpose of this study is to evaluate the use of this medication to prevent or reduce attacks and symptoms in those with an acute porphyria (AIP, HCP, VP, ADP). When enrollment opens, the APF staff will contact you with the name and phone numbers of the participating centers near you. If you are interested in participating for this exciting new trial, call the APF TODAY at 713.266.9617 or 1.866.APF.3635 with your name, porphyria type and location to be put on the list TODAY!
Remember.Research is the key to your cure!
Edrin R. Williams, MHSA
Part 3. Not Understanding the Difference Between Laziness, Leisure, and Need Prevents Care
Friday - November 17, 2017 @ 07:00:03
3. Not Understanding the Difference Between Laziness, Leisure, and Need Prevents Care
No one helps, and everyone tends to be skeptical about invisible illness, because they assume laziness is more likely. Especially here in the United States, where being weak, injured, or otherwise unable to work is viewed as despicable.
If we are each the masters of our own fate, then no one should have to take care of us and nothing should be given to us for free.
Just try going through the social security process or trying to keep a job with an illness that leaves you unable to move or function for days, for weeks, for months.
Worse than that, many of the services disabled people need are viewed as luxuries, as leisure activities. If youre rich or well off, its acceptable to get regular massages, to take vacations, to have housecleaners and in-home nannies. You might be made fun of for it, but its not considered unusual.
Yet when my fibro keeps me from doing basic chores or shopping or dealing with strict work schedules, I am looked upon with scorn and disbelief.
Most people have no idea what its like to be chronically ill. They take their health for granted. They view it through their lens instead of through our eyes.
And of course, if they did what we did or, rather, what little were able to it would be because they were being lazy or seeking out a luxury.
In-home care, service animals, and pampering activities are often necessities for people who have painful chronic illnesses. Its the only way to lessen our symptoms and be able to have enough spoons (energy) left over to take care of ourselves.
We are not and never will be healthy.
But wed like to get as close as we can. That involves those services and relaxation activities that everyone else takes for granted. Because while we have some good days, that doesnt mean were actually feeling better.
Which actually leads right into the next problem.
Part 2. Treating People With Chronic Illness Like Theyre Faking It Is Really Harmful
Wednesday - November 15, 2017 @ 13:57:47
2. Treating People With Chronic Illness Like Theyre Faking It Is Really Harmful
Thats not a hyperbole.
My misdiagnosis with Bipolar Disorder resulted in my being medicated with a substance I was highly allergic to. I ended up having seizures after the first dose. It scared me so much that I didnt seek any sort of treatment for a couple more years.
Meanwhile, my endometriosis got worse, eventually leaving me unable to attend classes or work. I was bedridden, without the energy to fight for my place in college, and unable to explain to any of the administration why I was ill.
Without the understanding of how my health affected my ability to attend, I had no recourse or strength to fight back when they decided to end my time at college. Rather than working with me on a solution, I ended up with a mountain of debt, no degree, and no income.
Many people with chronic illnesses are given options that are difficult, irrelevant, or downright impossible for us.
Our actual capabilities arent taken into account. And people figure, if healthier people can do it, we should be able to.
But for those of us with invisible illnesses, being treated appropriately can mean the difference between life and death. Some of us are given the wrong medical care (or worse, unsympathetic care).
This lack of acknowledgement occurs in many ways. People dont think you really need that handicapped seat on the bus. They report you for having a handicapped sticker when clearly you can walk. They fire you from your job because theyve never heard of your illness.
Many of us are accused of faking our illnesses when we need to ask for certain things: like asking that places be fragrance-free, needing to leave a loud party, or needing to take three naps in one day.
People tend to try to help when we dont actually need it and then fail to offer assistance when we really need it. Paying attention, learning about illnesses, and listening to our experiences goes a long way to ensuring we can get the help we need.
Each chronic illness manifests in different ways. Sometimes were capable of certain things and other times were not. Checking in with us at each step along the way for any activity is best. Doing your own research on our conditions will give you a better understanding of our needs. Itll also help us to save our spoons for getting through our day.
In many cases, its healthier for us not to push through our pain. Its better not to force ourselves into uncomfortable situations.
And it certainly wont help anything to add on stress (no matter how little) to our day.
In the end, its better not to make assumptions that make our days and lives harder.
Porphyria Post
Wednesday - November 15, 2017 @ 07:00:33
Want your physician to become better educated about your disease?
Personally invite them to this important symposium!!!
2018 Heme Biosynthesis and the Porphyrias: Recent Advances
Orlando, Florida - January 12-14, 2018
This three day symposium will focus on the most recent findings on the molecular biology and regulation of heme biosynthesis and the clinical features, pathophysiology, and current treatment of the acute hepatic and erythropoietic porphyrias. Day 1 will be devoted to the clinical aspects and current treatments of each Porphyria. Days 2 and 3 will focus on the latest research findings on heme biosynthesis and emerging molecular-based treatments for the porphyrias. Also, there will be a special session for patients on the latest advances in the Porphyrias.
This symposium is open to all healthcare professionals including clinicians, researchers, nurses, genetic counselors, Junior Faculty, Pre/Post Doctoral students, fellows and medical students.
The Symposium offers up to 25.5 CME Credits. The Registration fee includes meals and the poster reception. Orlando has an international airport and outstanding attractions such as Disney World and Universal Studios. The hotel rates are attractive ($133/night plus tax, single or double), with parking, Internet, and resort fees included. The room rate is available for three days after the symposium, on a first come basis.
To share this information with your doctor, you can provide them with the registration link or print and share the attached brochure and take to your appointments with you: www.porphyriassymposium.com
Part 1 Chronic Illness Has Unexpected, Horrible Side Effects
Monday - November 13, 2017 @ 07:00:17
1. Chronic Illness Has Unexpected, Horrible Side Effects
Unlike popular medical shows like House, many people with an invisible illness can actually have more than one. Its like winning the worlds worst lottery.
The first illness I was diagnosed with was endometriosis. Then the doctors realized I had PCOS (polycystic ovarian syndrome). Then due to my stressful life circumstances, my traumatic history of sexual and emotional abuse, my intense work schedule, and the fact that my body was so used to being in pain one day I woke up in an immense amount of pain that simply never went away.
Fibromyalgia: the nervous system literally operates on 100%, constantly engaging the flight or fight response until all it can do is turn on itself, electrocuting you from the inside, causing body-wide muscle aches, disrupting your temperature, making all of your senses even more sensitive, and leading to chronic fatigue and brain fog.
Thats not all it does; thats just the beginning.
Yet this is the actual slippery slope of chronic illness. If you have one already, youre more likely to develop others.
I didnt just develop full-blown fibromyalgia; I also found myself depressed and anxious from dealing with each of my physical illnesses and painful history. My PCOS alters my hormone levels, which leads to cyclic depression that matches my menstruation. Add that to the PMDD, and you have my very many suicide attempts.
Pregnancy and menstruation are dangerous for me in ways most people dont realize. When Im unable to receive the care I need and when others belittle or dont understand what Im going through, it can lead to isolation, depression, and anxiety.
All of my symptoms worsen when life circumstances are also stressful. In societies where simplicity is an ideal, the complexities of living with a chronic illness that no one else can see can make life that much harder.
Sometimes, it can even lead to death.
When you dont realize that its never just one thing affecting our health, but everything, then you can better understand our fatigue, our depression, why we say we cant do certain things.
Youll understand that pressuring us about work, education, or other normal activities only adds to our stress, which makes every part of our illness worse.
6 Things You Need to Know About Invisible Illnesses Part 1 of 6
Thursday - November 9, 2017 @ 07:00:05
6 Things You Need to Know About Invisible Illnesses
Even though Porphyria is not mentioned here, most if not all of us can understand these 6 Things of Invisible Illness Part 1 of 6
Imagine being so frozen with fear and dread that you refuse to get out of your car.
Even though you know youre sick, you know as soon as you step out that people will see your lack of a wheelchair or crutches and do a double take of the handicapped spot youve parked in.
The sad truth is that unless your disability is blatantly physically obvious, able-bodied people assume youre just like them.
Yet the truth is that about 96% of us disabled people have what are called an invisible illness. And because theyre invisible, people assume were faking all the time. And the scariest thing is that many painful chronic illnesses go without acknowledgement or recognition from society, even within disability circles.
An invisible illness can be mental, physical, or both. There usually arent too many outward signs of said illness, which is why the phrase but you dont look sick is both widespread and completely missing the point.
Take me, for instance.
I look like a rather healthy and young person. And yet, I have maybe ten invisible illnesses. Starting with my very first menstrual cycle, I spent two weeks out of every month doubled over in pain, wanting to kill myself, being told it was all in my head.
I grew older and eventually the intense pain spread to the rest of my body. And as the sexual and emotional abuse worsened, so did my mental health. Eventually I became a bit of a shut in.
Then I began receiving worse and worse diagnosis of chronic invisible illnesses.
The most debilitating ones are endometriosis, Polycystic Ovarian Syndrome, Premenstrual Dysphoric Disorder, and fibromyalgia. I dont suffer as much from my Post-Traumatic Stress Disorder anymore, but I do still deal with secondary depression and anxiety.
Most of these are things people have never heard of, or ones that people didnt realize could apply to people like me.
Most people lack the information or experience to empathize with our struggles. So here are six experiences you may never have realized people with chronic illness have to deal with.
Tom Collier A Long Road to Diagnosis EPP
Tuesday - November 7, 2017 @ 06:30:04
Tom Collier: A Long Road to Diagnosis EPP Conference Call For EPP patients it would be wonderful to be able to withstand and even enjoy more time in the outdoors, and to gain potential benefits for liver health. Being able to better avoid flare-ups from EPP is an important treatment goal, as Dr. Bloomer and Dr. Roth both noted that the main treatments available for the pain of EPP symptoms are aspirin, Tylenol (acetaminophen) and cold compresses. Dr. Bloomer encouraged everyone interested in the trials to stay in touch with the APFwe will let you know as soon as we have concrete information about afamelanotide trials. Almost all of the call participants wanted to know if they will be able to participate in afamelanotide trials and when the trials will start. Here we summarize some of the remaining issues addressed during the calla full account is on our website. â?¢ Liver disease. Dr. Bloomer stressed that very few people with EPP will develop liver disease, and that the best way to prevent liver disease is through vigilance and prevention: protect your skin carefully and have your doctor check your protoporphyrin levels at least once a year. â?¢ Surgical lights. Dr. Bloomer said it is absolutely necessary to cover surgical lights with UV protective films in EPP liver transplant surgery to prevent burns to the skin and internal organs. It is good caution to use the same films in other surgeries on EPP patients as well. â?¢ Drug safety in EPP. Both doctors stressed that EPP patients should avoid drugs, like estrogens, that are known to block the flow of bile. But Dr. Bloomer cautioned against confusing the medication issues in EPP with those in the acute porphyriasthe drug precautions for acute porphyrias do not apply in EPP. â?¢ Donating blood, organs and bone marrow. EPP patients should absolutely not donate bone marrow, to avoid giving EPP to the marrow recipient. Donating whole blood and organs other than the liver should be safe. â?¢ Current recommendations for the annual blood tests for EPP patients are on our website at www.porphyriafoundation. com/about-porphyria/types-of-porphyria/EPP ~ Dr. Roths recommendations for an EPP medic alert bracelet: www. brighamandwomens.org/eppref/Patient/medicalert.aspx ~ For UVF protection, Dr. Roth recommended Llumar auto glass film: www.llumar.com/en/Automotive.aspx ~ COTZ Total Block (with Titanium Dioxide and Zinc Oxide), available from sunprecautions.com ~ coolibar.com and others, as sunblock; and mentioned the Solumbra line of clothing from www.sunprecautions.com 65077 XT , not suo H, 087 eti uS, ya wdoo W0094 det seuqer eci vr es sser ddA The information contained on the American Porphyria Foundation (APF) Web site or in the APF newsletter is provided for your general information only. The APF does not give medical advice or engage in the practice of medicine. The APF under no circumstances recommends particular treatments for specific individuals, and in all cases recommends that you consult your physician or local treatment center before pursuing any course of treatment. All information and content on this Web site are protected by copyright. All rights are reserved. Users are prohibited from modifying, copying, distributing, transmitting, displaying, publishing, selling, licensing, creating derivative works, or using any information available on or through the site for commercial or public purposes. porphyriafoundation.org
Porphyria Post Important News
Wednesday - November 1, 2017 @ 08:31:44
PORPHYRIA POST - Important News
PATIENT MEETING - Atlanta, GA
Members in the Atlanta, GA area are invited to attend a Patient Education and Support Meeting. There will be a presentation and question/answer session with an on-site Porphyria expert (Dr. John Phillips). Come meet old and new friends who share your experiences with Porphyria. Please RSVP to Edrin Williams in the APF office at 1.866.APF.3635 oredrinw@porphyriafoundation.org. We look forward to seeing you there! Please see the invite below for additional meeting details.
SET YOUR DVRs - Out of the Shadows
Emmy nominated Dateline NBC documentary "Out of the Shadows" will re-air on Wednesday, November 8, 2017 at 4:00 AM. Please set your DVRs to watch this important documentary featuring EPP.
You can also watch the full "Out of the Shadows" episode here:
The Cook family is preparing to put on another fantastic barrel race. As you may know, the Cook brothers, Cason and Caul, have EPP and have set a great example about enhancing awareness of the disease in their community. The shadow race will be held in their hometown of Vernon, Texas. All proceeds will benefit the American Porphyria Foundation.
2018 HEME BIOSYNTHESIS AND THE PORPHYRIAS: RECENT ADVANCES SCIENTIFIC CONFERENCE and CLINICAL DAY FOR PATIENTS!
Mark your calendars to join the American Porphyria Foundation and the Genetic Disease Foundation along with expert physicians from around the world!
January 12-14, 2018
Click on the link below to view the agenda and register for Clinical Day for Patients. We look forward to seeing you there. If you have any additional questions, please contact the APF at 1.866APF.3635.
The Alnylam Trials for Givosarin will begin soon. The purpose of this study is to evaluate the use of this medication to prevent or reduce attacks and symptoms in those with an acute porphyria (AIP, HCP, VP, ADP). When enrollment opens, the APF staff will contact you with the name and phone numbers of the participating centers near you. If you are interested in participating for this exciting new trial, call the APF TODAY at 713.266.9617or 1.866.APF.3635 with your name, porphyria type and location to be put on the list TODAY!
"Remember.Research is the key to your cure!"
Welcome November Tips for Holiday Travel & Nathan Wayne Carr HCP
Friday - October 27, 2017 @ 07:00:19
Welcome November 2017
Please give a warm welcome as we have a new Greeter Nathan Wayne Carr. Nathan is a gentle and kind man with much experience in his HCP Porphyria.
As we enter into busy times these next few months, make sure to slow down, rest, proper steps for nutrition & hydration. If your traveling do you have all your medications with you in a safe place, let your friends and family know where you will travel to.
If you have an Emergency with your health do you have your ER Kit with you, a patient packet, or send your Dr an comprehensive Dr Kit? If not and you need assistance please call the APF @~1/866/APF/3635.
We will work with you quickly to assist you.
porphyriafoundation.org
Sit back and enjoy getting to know Mr. Carr and his experience of HCP.
PORPHYRIA POST: Health Professional Radio interview
Wednesday - October 25, 2017 @ 19:06:28
PORPHYRIA POST: Health Professional Radio interview
Desiree Lyon Howe, APF Executive Director, was given the opportunity to enhance porphyria awareness by participating in a live radio interview with Health Professional Radio. Desiree spoke on the acute hepatic porphyrias, raising patient and physician education, misdiagnosis and much more.
Health Professional Radio is an online radio/podcast network targeting 35K global healthcare professional listeners with a focus on "news and talk" geared toward a range of healthcare professionals across specialties and with a range of clinical experiences (including residents, nurses, and MDs). The show includes interviews with experts and everyday people on important health topics; some content is clinical, some management orientated, and some lifestyle orientated.
Alnylam is currently conducting a study with individuals who have acute hepatic porphyria (AHP), including AIP, VP, HCP, or ADP, as well as with caregivers of someone with AHP. The purpose of this study is to receive feedback about a newly developed porphyria electronic diary. Please click here to open a document for more information about this study.
Please note Its important that the participants in this research are not identified to Alnylam. If you are interested in participating, please contact:
A phone screen will take place to ensure that eligibility criteria are met before an in-person interview is scheduled. Participants must provide genetic documentation of AHP and have had at least one attack in the last six months. The interview will be conducted in-person, will be approximately 60 minutes long, and will be audio-recorded. Participants will be compensated for their time. Alnylam will not know the names of any participants, and the feedback we receive will be anonymized.
Warm Regards, Alnylam Pharmaceuticals
Liver disease in erythropoietic protoporphyria: insights and implications for management
Tuesday - October 24, 2017 @ 11:09:59
Liver disease in erythropoietic protoporphyria: insights and implications for management
This article has been cited by other articles in PMC.
The porphyrias are a group of disorders caused by defects in haem biosynthesis (fig 11).). Of the seven main types of porphyria recognised, two are characterised by associated liver disease (table 11).). In porphyria cutanea tarda it is the liver disease which leads to the onset of the porphyria, characterised by blistering, hirsutes and skin fragility of sunexposed skin. A number of different liver diseases may precipitate porphyria cutanea tarda including haemochromatosis, alcoholic liver disease and hepatitis C. In contrast, in erythropoietic protoporphyria (EPP) it is the porphyria itself which leads to liver disease, due to progressive deposition and accumulation of insoluble protoporphyrin IX in hepatocytes and bile canaliculi.
Figure 1The haem biosynthetic pathway showing the enzyme deficiency associated with porphyria cutanea tarda (PCT) and erythropoietic protoporphyria (EPP). The final step in this pathway involves the incorporation of iron into the middle of the
Table 1Liver disease and the porphyrias: names and patterns of inheritance for the seven main clinical variants of porphyria, highlighting those characterised by concomitant liver disease
EPP is an inborn error of haem biosynthesis caused by mutations in the gene encoding the mitochondrial enzyme ferrochelatase (FECH), the final enzyme in the haem biosynthetic pathway (fig 11).1,2,3,4,5 It was first described by Magnus et al in 1962.6 Ferrochelatase catalyses the insertion of ferrous iron into protoporphyrin to form haem, and when defective or deficient, accumulation of protoporphyrin ensues. Ferrochelatase is active in cells that produce haem including erythroid precursors in the bone marrow7 and hepatocytes.8 However, the majority of protoporphyrin (80% or more) originates from bone marrow with most of the remainder generated by the liver (fig 22).7,9
Figure 2The fate of protoporphyrin IX in erythropoietic protoporphyria.
Protoporphyrin accumulates in the maturing red blood cells during haematopoiesis. When red cells enter the circulation, free protoporphyrin diffuses across the red cell membrane and binds to plasma proteins. The liver extracts protoporphyrin from the plasma, most of which is excreted unchanged into the bile, with the remainder metabolised (by liver ferrochelatase) to haem. Some protoporphyrin is subsequently reabsorbed in an enterohepatic circulation.10
Protoporphyrininduced hepatotoxicity is a rare complication occurring in 15% of patients, for whom liver transplantation is often required. Since the first liver transplant for EPP in 1980,11 more than 40 further liver transplants have been carried out as treatment for advanced liver disease in this condition. However, liver transplantation fails to correct the underlying metabolic deficiency and protoporphyrin damage to the transplanted liver is likely.
EPP is an inherited disorder with both recessive12,13,14,15,16 and dominant patterns of inheritance (fig 3A, BB).). In most patients with EPP, diseasecausing mutations are present on one allele in association with coinheritance of a lowexpression allele.17 This has been demonstrated by casecontrol association in 39 families with EPP.18 Using haplotype segregation analysis, a polymorphism was identified in intron 3 (IVS348C) that increases the use of an aberrant splice site.19 The aberrantly spliced mRNA has been shown to be subject to more rapid degradation resulting in a decreased steadystate level of mRNA, leading to a further reduction in FECH enzyme activity and disease expression.19 The lowexpression variant IVS348C has a prevalence in the white population of France of about 10%.18 Coinheritance of a FECHmutation and the lowexpression allele accounts for nearly all cases of expressed EPP,20,21,22,23 with estimated true autosomal recessive inheritance accounting for about 3% of cases.23 Very rarely, alternative mechanisms may reduce FECH activity below the critical threshold for symptomatic disease, including deletion of an FECH gene secondary to leukaemia24 or a dominant negative effect from the mutant FECHallele.25
Figure 3Genetics of erythropoietic protoporphyria (EPP) and relationship between genotype and ferrochelatase activity (expressed as a percentage of normal). (A) Autosomal recessive inheritance. (B) Autosomal dominant inheritance due to coinheritance
The FECH gene was cloned and sequenced in 199026 and subsequently localised to the long arm of chromosome 18 (18q22.31).27 It spans 45kb and contains 11 exons which code for an enzyme with 423 amino acid residues.28 The enzyme functions as a dimer, which may have reduced stability and catalytic activity in the presence of a mutated subunit.29 Allelic heterogeneity of the molecular defects in the FECHgene has been demonstrated.30 Analysis of the genetic mutations in EPP reveals three main categories:
Nucleotide substitutions: missense and nonsense mutations caused by single nucleotide substitutions in the coding region; nonsense mutations are null.
Splice site mutations: these may produce truncated proteins but this has never been directly shown for EPP. Furthermore, these mutations do not always produce stable mRNA transcripts; they may be null or may preserve some activity.
Frameshift mutations resulting in premature stop codons are always null, the mechanism being accelerated RNA decay.
Minder et al described a significant genotypephenotype correlation between socalled null allele mutations and protoporphyrinrelated liver disease in EPP.31 This supported an earlier report which showed major structural alteration in the FECH protein in all of eight cases undergoing liver transplantation for EPPassociated liver failure.32,33 However, as more data accumulate, it is increasingly clear that the FECHgene mutations by themselves do not account for the severe liver disease phenotype, as the same mutations have now been reported both in asymptomatic family members and in patients from families in which liver disease had not occurred.33,34 There is currently no way reliably to identify patients at risk, and no intervention that is uniformly effective in restoring normal liver function once hepatic failure ensues.
No study to date has specifically set out to document the natural history of liver disease or to identify risk factors in its causation in a large unselected cohort of unrelated patients with EPP. The largest studies reported so far have all been from single centres and many are subject to case selection bias resulting from local interest and expertise in the management of EPPrelated liver disease. Despite these shortcomings, these studies (summarised in table 22)) provide a useful starting point for further analysis of this topic.
Table 2Patient series with erythropoietic protoporphyria (EPP) worldwide with details of liver disease
Irrespective of its origin, excess protoporphyrin is excreted by the liver into bile and enters an enterohepatic circulation. Protoporphyrin is a hydrophobic compound which is not filtered by the kidneys. When in excess, protoporphyrin becomes insoluble in bile and exerts cholestatic effects leading to architectural changes in the hepatobiliary system ranging from mild inflammation to fibrosis and cirrhosis.42 Even in early EPP, ultrastructural damage has been described in hepatocyte nuclei, endoplasmic reticulum, plasma membranes and bile canaliculi, associated with protoporphyrin crystals.43Significant intracellular precipitates of protoporphyrin, demonstrated in liver biopsy samples by fluorescent birefringence, are invariably present in protoporphyric liver disease.44
Exposure of cultured hepatocytes to protoporphyrin inhibits cell metabolism and increases cellular fragility.45 However, it remains unclear what effect protoporphyrin has on hepatocytes in vivo and how this relates to the development of liver disease. Bloomer et al32 found that liver FECH activity in EPPrelated endstage liver disease was reduced more than could be explained by the decrease in ferrochelatase protein, and concluded that the liver probably contributes to the overproduction of protoporphyrin that results in its own damage. In the absence of a clear explanation for occasional severe liver disease complicating EPP in humans, Nordmann46 speculated that patients may vary in their susceptibility to protoporphyrininduced liver damage. This is probably so, but host factors other than deficiency of FECH activity relevant to the onset and progression of liver disease are currently unknown. Recent murine studies (highlighted later in this review) have revealed that other genetic factors are relevant to this process. It is likely that quantitative trait loci analyses will shed more light on this important topic, with the growing recognition of the importance of such factors for many inherited diseases.
Hepatobiliary disease in humans with EPP may be described under the following headings:
Cholelithiasis;
Mild liver disease;
Deteriorating liver disease; and
Terminal phase of EPPassociated liver disease.
Cholelithiasis
Protoporphyrin in bile may crystallise out forming stones. The original case of EPP described by Magnus in 1961 underwent a cholecystectomy at the age of 29years and a solitary gallstone was identified.6Gallstones have subsequently been reported in EPP in many patients, including 2 patients in a series of 29 from Denmark,35 4 patients in a series of 32 reported from the USA,36 and 9 patients from a series of 200 reported from The Netherlands.37 Three of the patients in the series from the USA required cholecystectomy, and gallstones analysed from 2 of these cases revealed high levels of protoporphyrin.36Todd highlighted the fact that, when gallstones occur in children, EPP should be included in the differential diagnosis.47
Mild liver disease
There is wide variation in the severity of liver disease in EPP. Minor abnormalities in biochemical parameters of liver function are relatively common and include raised aspartate transaminase levels and approximately twofold increases in alkaline phosphatase and γglutamyl transferase.44 A study of 32 patients with EPP included a single patient with abnormal liver function.36 Analysis of liver biopsies from this case and 4 patients with normal liver function revealed protoporphyrin deposition without evidence of fibrosis or infiltrates in all 5 samples.36 In contrast, liver biopsies from 7 cases of EPP without overt liver disease from The Netherlands showed protoporphyrin deposition and mild fibrosis in 3 cases; the remainder were normal.48 In a separate study Cripps et al also reported protoporphyrin deposition in liver biopsy specimens from 5 patients with EPP and normal liver function tests; portal and periportal fibrosis was identified in 2 of these 5 samples.49 An ultrastructural study of liver biopsy specimens obtained from 11 patients with EPP, 4 of whom had overt liver disease and 7 of whom did not, revealed significant pathological changes in all samples compared with normal controls.50 It was concluded that liver damage is an early and consistent feature of EPP.50 Finally, a study with histopathology and ultrastructural studies of liver biopsy samples obtained from 4 patients with EPP (1 with severe liver disease, 1 with mild liver disease and 2 without evidence of liver disease and with normal histopathology) showed characteristic crystalcontaining vacuoles on electron microscopy in all 4 cases.51 It therefore appears that protoporphyrin deposition in hepatocytes is invariable, whereas histological evidence of damage is less common; electron microscopy will, however, show ultrastructural evidence of damage in most, if not all, patients with EPP.
Deteriorating liver disease
Patients with EPP who manifest significant liver disease will progress to decompensated cirrhosis which, in the absence of liver transplantation, is fatal. Various treatments have been attempted to preserve liver function and break the cycle of rapid deterioration that occurs in this situation in order to avoid terminal liver failure, or at least to buy time until a donor liver becomes available. The different forms of treatment have been directed at specific pathogenetic mechanisms as follows:
To increase the excretion of protoporphyrin into bile by the oral administration of the bile salts chenodeoxycholic acid50,52 or ursodeoxycholic acid.41
To reduce protoporphyrin production by suppressing erythropoiesis using iron,53,54 red cell transfusions55 or infusion of haematin,56,65 all of which are intended to reduce the drive for haem synthesis.
To reduce the pool of circulating plasma protoporphyrin by plasmapheresis,57,58 haemodialysis,59and exchange transfusions.55,60
To reduce protoporphyrin levels by interrupting the enterohepatic circulation with administration of cholestyramine59,61 and activated charcoal.54,62
To reverse oxidative stress in EPP by intravenous vitamin E therapy.63
One or more of these treatments are sometimes combined,58,59 and this is currently the practice before liver transplantation in order to optimise the environment into which the new liver is transplanted.64,65However, none of these treatments is effective in all cases, each has potential problems and none has been applied in sufficient numbers of patients to allow a rigorous evaluation of efficiency.
Treatment with bile acid appears to have only a modest effect on EPPassociated liver disease. Administration of chenodeoxycholic acid resulted in no distinct improvement in ultrastructural assessment of organelle damage in EPPassociated liver disease in three patients after 1year of treatment,50 and its therapeutic efficacy in another study was doubtful.52 In spite of these reports, chenodeoxycholic acid continues to be used with other treatments for the treatment of acute liver decompensation before transplant surgery.64 Doss and Frank reported a patient who showed biochemical and clinical improvement from EPPinduced decompensated liver cirrhosis following treatment with cholic acid.66
The role of iron treatment in EPP is unclear, with reports of significant efficacy53,54 but also reports of increased protoporphyrin levels in some patients.67,68 Furthermore, use of erythropoietin following orthotic liver transplantation in one patient was implicated in causing a great overproduction in protoporphyrin IX, prompting the authors to conclude that treatment with erythropoietin is risky and probably contraindicated in EPP.69 Transfusion therapy is probably the most widely reported and effective treatment for deteriorating liver function in EPP,55,70,71,72,73,74 but in one case it was implicated as the trigger for worsening liver function.75 Various hypertransfusion protocols for decompensating EPP have been used, ranging from 1unit of blood per month for 5months to a maximum of 1unit every 27days repeated 310 times.55,70,71,72,73,74 However, care is needed as transfused cells exposed to plasma protoporphyrin are more fragile than endogenous protoporphyrinloaded erythrocytes.75,76,77 The risk of haemolysis can be reduced by plasmapheresis conducted before transfusion and, in the context of liver transplantation, immediately before surgery.65 Exchange transfusion is seldom used and is reserved for severe or rapidly deteriorating cases. Intravenous haematin has been shown to reduce protoporphyrin levels56,66,78,79 and, more recently, haemalbumin has been used successfully in combination with plasmapheresis before liver transplantation.65 Haemodialysis has only been used as a treatment in EPPrelated liver failure and was unsuccessful.59
The efficacy of oral cholestyramine was reported in two well documented cases.80,81 The therapeutic use of this agent in EPPrelated liver disease is seldom reported and, when used, it is usually in combination with other treatments.59,64 Some patients, however, fail to respond to this treatment.66 Cholestyramine was the main treatment used in a 36yearold patient with EPP who developed liver disease but remained in good health until rapid deterioration in liver function 6years later requiring liver transplantation.82Activated charcoal is another treatment aimed at preventing reabsorption of protoporphyrin from the gut, and has the merit of being cheap and safe, albeit unpalatable.54,62 Longterm treatment (27months) with this agent has been reported to be beneficial in reducing protoporphyrin levels and restoring liver function.62 Finally, intravenous vitamin E was reported to be effective at reversing severe EPPrelated liver disease in a single case report.63
Terminal phase of EPPassociated liver disease
Deteriorating liver disease in EPP is characterised by cholestasis83 followed by jaundice and generalised upper abdominal pain.66 The spleen becomes enlarged and haemolysis may ensue.75 Rapidly worsening photosensitivity due to a further reduction in biliary free protoporphyrin excretion heralds the onset of fulminant disease which is seldom reversible and, in the absence of liver transplantation, usually leads to death. Acute liver failure may rarely be the presenting feature for EPP.85 Additionally, EPPrelated liver failure may sometimes be further complicated by the development of acute pancreatitis.86 In 1986 Bonkovsky and Schned87 summarised 21 fatal cases of EPPrelated liver failure reported in the literature, and Todd identified a further 8 cases in his comprehensive review in 1994.3 The majority of these fatal cases were over the age of 30, but two teenagers and an 11yearold child were also included.3 In the last 10years liver transplantation has increasingly been available as a treatment option but, despite this, patients have continued to die from EPPrelated liver failure.30,59,88
The cycle of deterioration which characterises fulminant hepatic failure in EPP has been recorded in detail in a number of individual case reports, but the initiating event (or events) remains unclear. What is known is that cholestasis induced by protoporphyrin leads to further accumulation of protoporphyrin,45 initiating a vicious cycle of worsening cholestasis and reduced protoporphyrin excretion.45 Haemolysis leads to increased erythropoiesis, hence increased de novo porphyrin formation by the bone marrow.75 Once this cycle is established, liver decompensation is rapid and liver failure ensues ((figsfigs 4 and 55).).
Figure 4Liver disease in erythropoietic protoporphyria. An explanted liver showing black colour due to diffuse deposition of protoporphyrin pigment.
Figure 5Magnification Ã?20 of liver histology from fig 44 showing the birefringence of pigment deposits due to the presence of protoporphyrin crystals.
The first liver transplant for EPPrelated liver disease was carried out in 1980.11 Since then, more than 40 further liver transplants have been reported (table 33).). Published reports with clinical details of liver transplantation for EPPrelated liver disease include 41 patients (23 male) of age range 1359years (mean 39.2years). The most recent figures from the European Liver Transplant Registry indicate that 19 liver transplants were performed in Europe for EPP between 1985 and 2003, 13 of whom have survived. The reasons for the deaths were gastrointestinal haemorrhage (n=1), primary graft nonfunction (n=1), infection (n=2) and unknown causes (n=2) (V Karam, personal communication, July 2005).
Table 3Published clinical reports of liver transplants for erythropoietic protoporphyria (EPP)related liver failure
McGuire et al84 have recently reported the outcome of 20 cases of EPP in the USA who underwent liver transplantation. Paediatric and adult survival rates were 100% and 85% respectively at 1year, 75% and 69% at 5years and 50% and 47% at 10years. Recurrent EPP was noted in 11 of the 17 patients (65%) who survived more than 2months after transplantation. Of the remaining 6 patients without evidence of recurrent EPP, serial monitoring of liver function has shown no evidence of cholestasis. The earliest interval at which recurrent disease was noted on liver biopsy was 8months. Three patients were retransplanted for recurrent EPPassociated liver disease at 1.8, 12.6 and 14.5years. Three additional patients in this series died 6173months after liver transplantation, documented by extensive protoporphyrin deposits and bridging fibrosis or cirrhosis on liver biopsy. The high rate of recurrent EPPassociated liver disease prompted the authors to recommend that bone marrow transplantation (soon after successful liver transplantation) should be considered in transplant recipients in order to correct the underlying defect and prevent this.
Liver transplantation restores normal liver function and thus the ability to excrete protoporphyrin via the biliary system. However, it does not correct the FECH enzyme deficiency in the bone marrow, which continues to be the source of significant overproduction of protoporphyrin. Transplanted patients therefore usually continue to have the symptoms of EPP and are at risk of developing EPPrelated liver disease in the transplanted liver;41,89,90,104 some patients have subsequently required a second transplant.84
A number of early patients with EPP who received a liver transplant developed lifethreatening phototoxic abdominal burns and wound dehiscence with severe haemolytic anaemia, since it had not been foreseen that prolonged visceral exposure to operating lamps would result in tissue phototoxicity analogous to that displayed in the skin under normal circumstances.82,106 Additional and unexpected complications included acute neuropathy82,101,106 resulting from high circulating protoporphyrin levels (neuropathy is not generally a feature of EPP as it is of other forms of porphyria), and acute protoporphyrinmediated damage to the transplanted liver resulting in delayed return of function as a result of these high circulating levels at the time of grafting.41,89,90 This led to recommendations for optimising the environment for the transplanted liver,58 which included the use of filtered theatre lights and shortterm measures aimed at keeping the level of protoporphyrin as low as possible in the immediate postoperative period.90,102 The introduction of such measures has reduced perioperative complications including haemolysis.76Furthermore, longterm use of plasmapheresis and intravenous haemalbumin has been advocated as a worthwhile therapeutic measure to prolong survival of the transplanted liver in the face of chronically raised protoporphyrin levels.105 There are increasing reports of mediumterm30,103,105 and longterm104survival following EPPrelated liver transplantation. However, longterm followup of two patients with EPP who underwent liver transplantation for acute liver failure revealed protoporphyrin deposits and onset of fibrosis in the transplanted livers 8months and 6months after transplantation.104 Despite this, both patients remained in good health 7years after surgery.104
to continue reading this article click on this link: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1994365/
There are many different ways you can help with research, not just participating in a drug research trial. Below is one of the very different but equally important ways to make a difference:
Alnylam Pharmaceuticals is currently conducting a study with individuals who have Acute Hepatic Porphyria (AHP); AIP, VP, HCP, or ADP, as well as with caregivers of someone with AHP. The purpose of this study is to understand opinions about a newly developed porphyria Electronic Diary.
You or your caregiver can be a great assistance in this new study. It is important to know if this particular Electronic Diary is easy to use and efficient. The study is conducted by a vendor different from Alnylam. Please contact the APF if you would like to participate. Remember, your caretaker can participate, too. There are only 8 spots left, so join now.
Potential participants will be screened by phone and if they qualify, an interview will be scheduled. The interview will be conducted in-person, will be approximately 60 minutes long, and will be audio-recorded. Participants will be compensated for their time. Alnylam will not know the names of any participants, and the feedback received will be anonymous.
Porphyria cutanea tarda (PCT) is a form of porphyria that primarily affects the skin. People affected by this condition generally experience "photosensitivity," which causes painful, blistering lesions to develop on sun-exposed areas of the skin (i.e. the hands and face). Skin in these areas may also be particularly fragile with blistering and/or peeling after minor trauma. In some cases, increased hair growth as well as darkening and thickening of the affected skin may occur. Liver abnormalities may develop in some people with the condition and PCT, in general, is associated with an increased risk of liver cirrhosis and liver cancer.[1][2] In most cases, PCT is a complex or multifactorial condition that is likely associated with the effects of multiple genes in combination with lifestyle and environmental factors. For example, factors such as excess iron, alcohol, estrogens, smoking, chronic hepatitis C, HIV and mutations in the HFE gene (which is associated with the disease hemochromatosis) can all contribute to the development of PCT.[3][1] Less commonly, PCT can run in families (called familial PCT). Familial PCT is caused by changes (mutations) in the UROD gene and is inheritedin an autosomal dominant manner.[4][1] Treatment may include regular phlebotomies (removing a prescribed amount of blood from a vein), certain medications, and/or removal of factors that may trigger the disease.[1][2]
The Human Phenotype Ontology (HPO) provides the following list of features that have been reported in people with this condition. Much of the information in the HPO comes from Orphanet, a European rare disease database. If available, the list includes a rough estimate of how common a feature is (its frequency). Frequencies are based on a specific study and may not be representative of all studies. You can use the MedlinePlus Medical Dictionary for definitions of the terms below.
The resources below provide information about treatment options for this condition. If you have questions about which treatment is right for you, talk to your healthcare professional.
Management Guidelines
Orphanet Emergency Guidelines is an article which is expert-authored and peer-reviewed that is intended to guide health care professionals in emergency situations involving this condition.
The American Porphyria Foundation offers a document that includes information about porphyria, types, testing, and treatment with Panhematin. Click the "document" link above to view these guidelines.
Research helps us better understand diseases and can lead to advances in diagnosis and treatment. This section provides resources to help you learn about medical research and ways to get involved.
Clinical Research Resources
ClinicalTrials.gov lists trials that are studying or have studied Porphyria cutanea tarda. Click on the link to go to ClinicalTrials.gov to read descriptions of these studies.
Please note: Studies listed on the ClinicalTrials.gov website are listed for informational purposes only; being listed does not reflect an endorsement by GARD or the NIH. We strongly recommend that you talk with a trusted healthcare provider before choosing to participate in any clinical study.
Patient Registry
The Porphyrias Consortium is a team of doctors, nurses, research coordinators, and research labs throughout the U.S., working together to improve the lives of people with this condition through research. The Porphyrias Consortium has a registry for patients who wish to be contacted about clinical research opportunities.
Making decisions in health care and ethics. Why prepare? Prepare for the unexpected. It seems we often go through the motions of preparing for unexpected life events without really believing that they might happen. Investing the time or energy, or thought required to be adequately prepared for something serious illness is not always at the top of our to-do list. We may not have much money to put aside for a rainy day, thinking about life or health Insurance many of us may or may not have.
How often do we complete and sign off on these documents, only to file them away and forget about them with little, if any discussion with loved ones impacted by these decisions? Because some of these discussions are not easy, it is common to want to avoid them. I know i did. It was a good time to honestly sit down with my family, because in my case (AIP) my attacks were changing and increasing no matter what we have tried and with age other things in the body, honestly do not get any better. So no more excuses, its time to be honest and talk and think about the options and be prepared. The worst thing you can do is NOT BE PREPARED! Speaking from experience when one of my parents passed away unexpectedly, their was nothing really written and sealed as to their wishes, and after things, home, items what to do with them and how. It caused a lot of stress to take that upon and in my case having AIP and stress is a big trigger for me. That's why I want to talk about this. All of these preparations are important in ensuring that our choices and wishes are honored through-out life. It is very clear that preparation, however, without discussion can often be inadequate and incomplete. When sick with Porphyria we already feel defeated and are extremely sick or in so much pain we still have to be very alert and our own advocate. Confronted with multiple decisions regarding our healthcare, both patients and families can be overwhelmed. When discussions have been part of the preparation , it can ease the burden of decision making, reducing the stress for loved ones, especially when a crisis actually occurs. When we have the opportunity to share our health care goals, our wishes, values and that which gives us quality of life, with our loved ones and our professional caregivers, it becomes easier to make the necessary decisions if that time should arrive. Remember the importance of those difficult conversations and consider them a part of your health care discussions and decisions today. At first, I personally had to think about it, timing things out with my family members, choose the time. I had to do it swiftly but also ask, am I ready to discuss it? How will they react? If you have not already done so i encourage you to explore Advance Care Planning. It is a process to explore, understand and document your wishes for future health care if you become unable to do so. Seek out a health care professional today and ask for a Health care document, ask for a social worker to help explain the document its purposes and how it benefits you.
Preparing today will bring you, family and friends peace of mind.
Member story Ann Warnke on EPP
Friday - October 13, 2017 @ 13:50:25
Ann Warnke
Erythropoietic Protoporphyria (EPP)
Living with EPP Ann Warnke had her first symptoms of Erythropoietic Protoporphyria (EPP) when she was 13 years old. A friend of the family took Ann and some other kids deep-sea fishing, and after she got home that night, her face swelled up so much that she couldn't open her eyes and her nose disappeared. The capillaries under her skin burst too, so that she looked bruised as well as swollen. She says she looked "like I'd been beaten." Her doctors first thought the swelling and bruising were a bad reaction to something she had eaten, and Ann would go on to have many more photosensitive incidents before she was finally diagnosed with EPP as an adult. Yet the knowledge of her own diagnosis allowed Ann's younger son to be diagnosed by the time he was three years old. Just like Ann, Matthew got to go out on a boat for the day with his dad, but the sun made his skin burn to the point that holding a hot dog was painful, and his ears turned inside-out from the swelling. These days, both Ann and Matthew have their EPP well under control. They both use UV-protective clothing, and can even go skiing sometimes or take a cruise with the help of a face mask, an umbrella and pharmaceutical-grade beta-carotene. Ann notes that Matthew's diagnostic labs don't show such high numbers as her own, and that he appears to have milder photosensitivity than she does. But for both of them, a photosensitive reaction with EPP means swelling, itching and painful burning that can take five days to subside. During that time, the Warnkes turn the A/C down low, and use ice bags, Benadryl and Tylenol 3 to ease the pain. Ann uses Lumitene (pharmaceutical-grade beta-carotene) if she needs to increase her sun tolerance without increasing the symptoms of EPP. At home, she prefers to cover up and avoid spending time outdoors in the sun, because taken over a lengthy period Lumitene turns her skin orange and gives her some gastrointestinal upset. But when going on vacation she'll begin taking Lumitene a few weeks before leaving home, continue throughout the vacation, and then taper off it after she comes home. She still covers up all the time, but she is able to stand more time in the sun with Lumitene. Back home, Ann has special tinting on her windows to protect her from the sun's UV rays. APF Scientific Advisory Board member Dr. Micheline Mathews-Roth helped Ann with the window tinting and has gotten testing information to Ann's doctors. Ann calls Dr. Roth "just phenomenal, extremely good and accessible." Dr. Roth has been a wonderful resource both for the APF and for EPP patients. Ann is a wonderfully positive person and has a fantastic attitude about dealing with her condition.
Event Brite- FDA Scientific Workshop for Erythropoietic Protoporphyria (EPP) is now available for review
Wednesday - October 11, 2017 @ 12:37:28
A Message from United States Food and Drug Administration:
We are pleased to announce the publication of the summary report that summarizes the October 24, 2016 FDA Scientific Workshop on EPP. The report has been posted online and can be found here: https://www.fda.gov/downloads/Drugs/NewsEvents/UCM579234.pdf.
We are thankful for your participation and grateful to the patients, caregivers, experts and others who shared their perspectives at the workshop. For more information on this workshop, please visit the workshop website here: https://www.fda.gov/drugs/newsevents/ucm501389.htm.
Support Nicole & APF today: https://www.facebook.com/donate/120370012003528/
Jean-Baptiste Roberge Story of Erythropoietic Protoporphyria (EPP)
Saturday - October 7, 2017 @ 18:03:56
Jean-Baptiste Roberge
Type of Porphyria:
Erythropoietic Protoporphyria (EPP)
"Seizing the day even if it is raining outside".
Throwing a soccer ball into the goal has never been something easy to do for me. I am almost unable to catch a ball and throw it back without clumsily dropping it. When I was a kid, I never had the chance to go outside with friends and to simulate a football tactic. I just couldn't.
I still remember the summers in the countryside. I constantly needed to go back home to compulsively wash my hands. I still remember, as an 8 year old boy, trying to explain to a young day camp monitor that I could not play with the rest of the group on the beach. I still remember the judging look on my high school gym teacher's face when I was trying to explain that I should preferably stay in the shade of the trees while the others were scoring points on the soccer pitch. I still remember the feeling of guilt after having canceled a promising trip in Iceland, because I was terrified by a never ending sunset. I still remember the comments of my brothers telling me that all this was in my head.
Last summer, I heard about a hematologic condition. I read a lot on the subject, which tried to convince me that what I had was not that serious. Eventually, I met an internist and told him my hypothesis. A few extremely precise and unusual blood tests later, I met him again last January. On that day, the specialist was accompanied by an internal medicine resident. He told her to take advantage of this interview because I would, in all likelihood, be her only case of that condition in her entire career.
Because of my erythropoietic protoporphyria, which consists of an inherited enzyme alteration in the heme biosynthetic pathway, I have an accumulation of protoporphyrins in my red blood cells. These proteins abnormally react to visible and ultraviolet light, entering an excited energy state that damages my skin tissue and my liver and making me prone to gallstones and liver failure. Unfortunately, there is no effective way of lowering my circulating protoporphyrin levels. Beta-carotene, cysteine, carcinogenic occlusive sunscreens, and narrow wavelength phototherapy have been tried. Of all possibilities, a drug named, afamelanotide that acts to increase skin pigmentation by stimulating skin melanocytes has shown promise. It has been approved in Europe but not yet in the US. Lack of awareness for this rare disease slows down the regulators.
When I was a kid, I would have been so happy if at least one person could simply and fully understand me. Awareness for rare diseases such as erythropoietic protoporphyria could mean the world for people affected by them. Even if there is no cure available for my condition, I finally have a justification for my symptoms and a simple way to explain them to others without being judged in any way. As a future physician, I will do the very best I can to be aware of these rare and afflictive diseases.
Summers and their cruel sunbeams, winters and their sparkling snow: everything is against me. Everything? No! I have an ultimate compromise: avoiding the sunlight, wearing long shirts, walking away from the sun, planning on which side of the car I should sit during long trips, even resigning myself to never being very good at outdoor sports, being different, accepting this handicap, and still seizing the day, even if it is raining outside!!!
Go Nicole Castellano!
Friday - October 6, 2017 @ 07:00:25
We are so proud of you Nicole!
Congratulations and much success in raising awareness for the APF!
On October 8th 2017 I'm running in the 40th Anniversary Chicago Marathon which is quite a feat as just years ago I was in a wheelchair and couldn't walk due to the rare, devastating disease Porphyria. Porphyria is a rare genetic disorder that is incredibly painful and can be fatal. There is no cure for Porphyria YET. My goal is for us to raise $5,000, 100% of which will go directly to the American porphyria Foundation. Because porphyria is so rare it gets minimal funding from the government so any donations help immensely!
The American Porphyria Foundation advocates for patients all over the world and supports research to not only improve treatment but to one day find a cure. I'm so passionate about their Physician Education programs because I watched doctor after doctor year after year banging their heads against a wall trying to help me. Testing for porphyria IS available and the more the doctors know to look for porphyria, they can properly diagnose many more people and more quickly.
I believe one person can make a difference, please join me in being that one person and let's see what we can do together with the power of the ripple effect of social media honoring one of the most inspiring races in the world. Please donate at least a dollar and pretty please with sugar on top share this with your entire social media network and sincerely ask them to do the same thing to help the APF in its mission.
Press Release
Escaping from Deadly Porphyria to Running the Chicago Marathon, Nicole Castellano Inspires All!
Saint Charles, IL - On October 8 at the 40th Anniversary Chicago Marathon, Nicole Castellano will run to bring attention to a rare disease, Porphyria. Nicole is herself a survivor of this rare illness, with near fatal experiences that beat her down physically and mentally. At the marathon, every step she takes will help support others ailing with Porphyria and all donations received will go towards the American Porphyria Foundation (APF). Since Porphyria is rare, funding from the government is minimal, therefore donations are critical to increase awareness and research for a cure.
Nicole has seen it all and emerged triumphant. From a world class skating athlete, she was reduced to a shadow of her former self due to Acute Intermittent Porphyria, one of at least eight types of this rare disease. She became wheelchair bound unable to walk suffering burning, stabbing pain in her abdomen that was unbearable. She underwent nine abdominal surgeries, including a hysterectomy that wasnt required, that left her unable to have children. She experienced mental changes including hallucinations from a lack of oxygen in the brain that is common with Porphyria and almost died three times.
She says the worst part was not knowing WHY and living like that for 12 years before she was diagnosed and finally treated with the expensive drug Panhematin that she says saved her life. Nicole still gets monthly infusions through a port in her chest to keep Porphyria at bay.
An active role was played in Nicoles recovery by the American Porphyria Foundation, which is dedicated to improving the health and well-being of individuals and families affected by Porphyria. The APF was instrumental in doing research and getting the drug approved that Nicole and so many Porphyria patients rely on.
Before Porphyria attacked her body, Nicole attended Northwestern University 1991-95, graduating with honors with a double major in theatre and anthropology. Like any other teenager she dreamed about her future, and passionately took up photography and writing during a trip to Africa and even got her pilots license. She went on to start a moving and storage firm, and was awarded the 2005 Businesswoman of the Year by the Business Advisory Council. Her dreams were however soon to come crashing down with the advent of Porphyria.
A rare and difficult to diagnose disease, Porphyria inflicts chaos on the autonomic nervous system. Some symptoms are neurological problems, severe stomach and back pain, vomiting, nausea and sun sensitivity. Extreme pain and possible fatalities make Porphyria a dreaded illness which currently has no cure and is so rare it has Orphan Disease status.
Nicole hopes that since testing is available for Porphyria, the funds raised for the APF will go toward their Physician Education Programs so that doctors can diagnose more patients and more quickly so they dont have to live with the horrible uncertainty that tainted her recovery.
I began doubting everything I knew about myself and It affected my relationships, my self-worth and self-esteem. I stopped believing in myself, feeling like at times the doctors thought it was all in my head. I knew it wasnt, but I blamed myself for being sick, for not being able to function like I used to thinking that I should have been able to be stronger somehow. I now help other women see its not their fault theyre sick, theyre not alone and its not who they are, its just something happening to them right now, she says.
Nicole fought on bravely, started a painting business and an Arbonne Health and Wellness business, saying that her treatment got to her about 70% of her health back, but following Arbonnes specialized diet got her all the way to 100%. After attending an Arbonne leadership conference, she started dreaming again and clawing her way out of survival mode.
Just as the 40th Anniversary Marathon is a milestone for Chicago, this run marks a milestone for Nicole and her battle with not only Porphyria, but the stigma attached to it.
Nicole says, When youre fighting a rare disease its easier to have people that love you unconditionally help you through. Even if you dont have that, you can still bounce back stronger than ever and reclaim your happiness! It starts with taking control of your health and your life. I show women struggling with serious illness and relationships how to heal and get their life back, even when they think theres no hope.
Herrero C, To-Figueras J, Ingelmo M, Oliva R, Munoz C, Munoz E, Carreras C
Hospital Clinic i Provincial, Barcelona, Spain
Acute intermittent porphyria (AIP) is an autosomal dominant disorder with incomplete penetrance, caused by a deficiency of porphobilinogen deaminase (PBG-D) enzyme and characterised by acute neuropathic attacks. High prevalence is known in Sweden and in other areas of Europe. In Spain AIP occurs with very low prevalence but it may be underestimated. Material: Since 1992 fourteen patients with PAI attended the Hospital Clinic of Barcelona. We present the clinical, biochemical, enzymatic and genetic analysis of 8 of these patients and 17 close relatives. Methods: We analysed (a): urinary excretion of porphobilinogen (PBG) and delta-aminolevulinic acid (ALA); (b) erythrocyte PBG-D activity; (c) DNA analysis through SSCP and sequencing the PBG-D gene. Results: Increased PBG and ALA urinary excretion and decreased erythrocyte PBG-D activity were observed in all the AIP patients. Moreover, seven cases of asymptomatic AIP were found in the relatives by means of these techniques. Five PBG-D gene mutations have been identified. Three of the mutations detected in four of the families have been previously reported in other countries (730delCT in exon 12, 340insT and G11R in exon 7). Two of the mutations identified correspond to novel mutations; one of them is present in 3 of the 8 families studied (37.5%). A strong correlation between the presence of any of the mutations and a low PBG-D activity has been observed. Conclusions: The prevalence of AIP in Spain may be much higher than previously estimated. Therefore porphyrin, enzymatic and genetic analysis must be performed in the relatives in order to detect asymptomatic carriers. AIP in Spain is also a heterogeneous disease and two novel mutations have been identified, one of them could be particularly prevalent in this area.
"What I Wish I Knew"
Monday - October 2, 2017 @ 07:00:09
What I Wish I Knew
When I was first diagnosed with Acute Intermittent Porphyria I was initially shocked. I struggled with the news and then I pulled myself together. I experienced many different emotions and often felt I would somehow make sense of it and get past it as soon as possible. I wanted to power through it and get my life back again. I closed myself off to lots of support along the way believing I shouldnt ask for help or expose my weaknesses. When I look back on it know, I realize though I am coping the best way I know how, there were so many things I wish I had known that would have made it easier for me, and probably my family and friends to.
Knowledge is Power: What I wish I Knew
Im not going to talk to anyone about my Porphyria, I said
Have you ever said that?
Where can I Find Information
I felt lost, scared, emotional, could my family really understand?
Did they want to understand?
Did they believe me?
Did my Doctor even believe me?
Now that I understand what care is needed I asked myself?
Who am I going to find to Bring with ME? Do they want to go?
Will they support me.
Will they say I am crazy?
I have Never asked for Help Before!
Its too hard trying to communicate with my family, friends with phone calls and texts while Im so sick, why bother I asked myself?
Do I need a special group to go to? They will think Im nuts��
No Im just going to stay inside and sleep it off.
Is this stress, or anxiety, fear, am I going to die?
What am I going to do? Who will I turn to who will help me?
I often ask myself these things and its important to journal, talk to a trust friend and family member, pray, start searching and dont stop until you get an answer good or bad, learn to be patient and kind. Be firm when you should, be your own advocate! Teach, read, watch do all that you can so you can conquer correct your lifestyle do what you must do, so that at the end of the day you can say I have done my very best! Dont stop there. Start all over when you wake upâ?¦..
Amy L Chapman
Acute Intermittent Porphyria
I will survive!
Diabetes& Porphyria! Can You Reduce Your Risk?
Thursday - October 5, 2017 @ 13:22:43
For those who suffer from Diabetes and Acute Porphyria is becoming harder to control. Learn about some simple tips. I enjoyed reading this article and I think you will to.
DiabetesCan You Reduce Your Risk?
THE incidence of diabetes mellitus is increasing so quickly that it has become a global epidemic. There are two major types of diabetes. Type 1 starts mainly in childhood, and presently doctors do not know how to prevent it. This article is about type 2, which accounts for about 90 percent of all diabetes.
While in the past it was seen as exclusive to adults, more recently type 2 diabetes has also been affecting children. Experts claim, however, that the risk of type 2 diabetes can be reduced. A little knowledge of this insidious disease may prove helpful to you.*
What Is Diabetes?
Diabetes is a condition that causes a person to have an excessive level of blood sugar. The disease upsets the normal process of transferring sugar from the bloodstream into cells that need it for energy. The result is damage to vital organs and impairment of blood circulation, sometimes leading to toe or foot amputation, blindness, and kidney disease. A large proportion of diabetes patients die of heart attacks or stroke.
Excess body fat can be a major factor in type 2 diabetes. Experts believe that fat accumulated in the belly and waist may indicate a higher risk for diabetes. More specifically, fat in the pancreas and the liver appear to disrupt the bodys regulation of blood sugar. What can you do to reduce your risk?
Three Steps That May Reduce the Risk of Diabetes
1. Have your level of blood sugar tested if you are in a high-risk group. A medical disorder known as prediabetesa condition in which blood sugar is moderately higher than normaloften precedes type 2 diabetes. Both conditions are unhealthy, but there is a difference: Although diabetes can be controlled, it cannot yet be cured. On the other hand, some prediabetics have been able to bring their blood sugar back to normal levels. Prediabetes may have no obvious symptoms. Hence, this condition may go unnoticed. According to reports, about 316 million people around the world have prediabetes; yet, many of them do not realize it. For instance, in the United States alone, some 90 percent of prediabetes sufferers are unaware of their condition.
Prediabetes is not harmless, however. Besides being a precursor to type 2 diabetes, it has recently been linked to an increased risk of dementia. If you are overweight, not physically active, or have a family history of diabetes, you might already have prediabetes. A blood test can tell you if you do.
2. Choose healthful food. You might benefit from doing the following whenever it is possible and practical: Eat smaller portions than usual. Instead of sugary fruit juice and carbonated beverages, drink water, tea, or coffee. Eat whole-grain bread, rice, and pastain moderationrather than refined foods. Eat leaner meats, fish, nuts, and beans.
3. Stay physically active. Exercise can lower your blood sugar and help you maintain a healthy weight. Swap some TV time for exercise time, recommends one expert.
You cannot change your genes, but you can change your lifestyle. Doing what we can to improve our health is worth the effort.
Nicole Castellano went from living her life as a competitive athletic World Champion figure skater.to coping with three near death experiences related to her Acute Intermittent Porphyria. With proper treatment and a specialized diet, Nicole bravely rebuilt her life and has moved from a wheelchair to training for her first marathon! 26.2 is a lot of miles and we wish her the best of luck every step of the way.
Please support Nicole as she prepares to race in the Chicago Marathon on October 8th, 2017. Each mile will be a reminder of all she has done to overcome living with AIP. And every step of her run will support all who live Porphyria.
EVER WONDER HOW SCENESSE WORKS TO TREAT EPP? HERE IS AN EXPERIMENT TO HELP EXPLAIN
Dr. Jasmin Barman, a scientific advisor with the Swiss Society for Porphyria, created an experiment to simulate what happens when Scenesse is used to treat Erythropoietic Protoporphyria. In this video, Dr. Barman shows the experiment to APF member JT Von Seggern. Here is what happens:
· JT is holding a vial of protoporphyrin solution, the substance found in the blood of a person with EPP.
· The blood of an EPPer glows, but that effect is only visible under a special microscope.
· The protoporhyrin in the vial starts to glow when it is exposed to visible light. This is what happens inside the red blood cells of a patient with EPP.
· In the body, this results in second degree internal burns inside the veins.
· In this experiment, a filter made of yellow cellophane is placed between a blue light and the protoporhyrin substance. The solution stops glowing!
· The filter prevents the light from reaching the substance in the same way that Scenesse stops the sun from reaching blood.
Thank you to Dr. Barman and to JT for sharing this experiment with us to help explain the life-changing affect that Scenesse can have on a person living with EPP. Dr. Barman created this experiment to help both children and adults understand EPP.
WATCH THE VIDEO HERE:
VOLUNTEERS NEEDED FOR THE AMERICAN SOCIETY OF HEMATOLOGY CONVENTION IN ATLANTA!
The American Porphyria Foundation will host an exhibit at the 59th ASH Annual Meeting and Exposition. The meeting is located in Atlanta, GA from December 9-12, 2017. We are looking for volunteers to help host the exhibit - it will be prepared with furniture and educational materials. Volunteers will need to represent the APF - and will be prepped in advance. Please contact Edrin at the APF office if you live in the area and are willing to help support us at this event. APF Office: 1-866-APF-3635
IMPORTANT: EPP MEMBERS
"We need your help in our campaign to approve Scenesse."
If you or someone you know is in the EPP MEDICAL community (Doctor, RN, or Other Medical Professional) and HAS EPP to please contact Kristen at the APF. Contact her by email atKristen@porphyriafoundation.org or give her call at 301.461.9889.
Acute Intermittent Porphyria (AIP) is an ultra-rare disease that attacks the neurological system.
Nicole Castellano went from living her life as a competitive athletic World Champion figure skaterâ?¦.to coping with three near death experiences. With proper treatment and a specialized diet, Nicole bravely rebuilt her life and has moved from a wheelchair to training for her first marathon!
Please support Nicole as she prepares to race in the Chicago Marathon on October 8th, 2017. Each mile will be a reminder of all she has done to overcome living with AIP. And every step of her run will support all who live Porphyria.
The American Porphyria Foundation (APF) is dedicated to improving the health and well-being of individuals and families affected by Porphyria. To learn more about Porphyria please visit: http://porphyriafoundation.com
Nicole has AIP (Acute Intermittent Porphyria) an Acuteâ?¦
YOUTUBE.COM
Are YOU To BUSY?
Friday - September 22, 2017 @ 11:47:54
Do you feel you are too busy? If so, you are by no means alone. Everybody, everywhere seems to be busy, reports the magazine The Economist.
IN A 2015 survey of full-time workers in eight countries, many respondents said that they find it hard to meet the demands of both their work and their homelife. Causes included increased responsibilities at work or at home, rising expenses, and longer working hours. In the United States, for example, full-time employees report working an average of 47 hours a week. Nearly 1 in 5 claimed to work 60 hours or more!
In another survey, this one involving 36 countries, over one quarter of the respondents said that they often felt rushed even in their leisure time! Children too can be affected if overloaded with tightly scheduled activities.
When we constantly try to do more than time may allow, we can become stressedvictims of what has been termed time pressure. But is it possible to live a more balanced life? What role do our beliefs, choices, and goals play? First, consider four reasons why some try to do too much.
1 THE DESIRE TO PROVIDE WELL FOR ONES FAMILY
I was involved in my work seven days a week, says a father named Gary. I did it because there was always something better that I wanted to give my kids. I wanted them to have the things I never had. Despite their good motives, parents need to examine their priorities. Some studies suggest that both adults and children who attach a lot of importance to money and material possessions tend to be less happy, less satisfied with life, and less healthy physically than those who are not materialistic.
Children who are raised with an emphasis on material things are actually less happy
In an effort to position their children for future success, some parents overschedule both their children and themselves with various activities. Such well-meaning parents, says the book Putting Family First, are acting like recreation directors on a turbo-charged family cruise ship.
2 THE BELIEF THAT MORE IS BETTER
Advertisers try to convince us that we are depriving ourselves if we dont buy their latest products. Says The Economist: The explosion of available goods has only made time feel more crunched, as consumers struggle to choose what to buy or watch or eat in the limited time they have available.
In the year 1930, a leading economist predicted that technological advancements would give workers more leisure time. How wrong he was! Instead of quitting [work] early, observed Elizabeth Kolbert, staff writer for the New Yorker magazine, people find new things to needand these things cost money and time.
3 TRYING TO SATISFY THE EXPECTATIONS OF OTHERS
Some employees work grueling hours to avoid displeasing their employer. Coworkers can also exert pressure by making others feel guilty if they do not stay late. And then there is economic uncertainty, which can make people more willing to work longer hours or to be constantly on call.
Similarly, parents can feel pressured to conform to the hectic pace of other families. If they do not conform, they may feel guilty about depriving their children.
4 THE PURSUIT OF STATUS AND SELF-FULFILLMENT
Tim, who lives in the United States, says: I loved my work, and I worked at full throttle all the time. I felt that I had to prove myself.
Like Tim, many feel a strong connection between their self-image and their pace of life. The result? Busyness has acquired social status, says Elizabeth Kolbert, quoted earlier. She adds: The busier you are the more important you seem.
LEARN TO BE BALANCED
Leading a balanced life is good for our mental and physical health. Is it really possible, though, to cut back or slow down? Yes. Consider four suggestions:
1 CLARIFY YOUR VALUES AND GOALS
It is normal to want a measure of financial security. But how much money is enough? What constitutes success? Is it measured merely by income or material assets? Conversely, having too much rest or recreation can also increase time pressure.
Tim, quoted earlier, says: My wife and I took a hard look at our life and decided to simplify it. We made a chart that showed our current situation and our new goals. We discussed the effects of past decisions and what we would need to do to reach our goals.
2 REDUCE THE INFLUENCE OF CONSUMER CULTURE
Advertising can fuel such desires, pushing a person to work long hours or to indulge in excessive or costly recreation. True, you may not be able to avoid all ads. But you can limit your exposure to them. You can also carefully consider what you actually need.
Keep in mind, too, the power that your associates can have on you. If they avidly pursue material things or if they measure success in material terms, you may be wise to seek out friends who have better priorities. The one walking with the wise will become wise,
3 SET LIMITS ON WORK
Speak to your employer about your work and your priorities. And do not feel guilty about having a life away from your job. The book Work to Live says: Those who put up boundaries between the job and home or take vacations find one consistent revelation: There is no apocalypse while youre gone.
Gary, quoted earlier, was financially comfortable, so he decided to reduce his working hours. I talked with my family and suggested that we simplify our lifestyle, he said. Then we gradually took steps to do so. I also approached my employer with a proposal to work fewer days each week, and he agreed.
4 MAKE FAMILY TIME A HIGH PRIORITY
Husbands and wives need to spend time together, and children need time with their parents. So avoid trying to match the pace of other families who are constantly on the go. Declare some downtime, Gary suggests, and drop things that have a lower priority.
When your family is together, do not let television, cell phones, or other devices isolate you from one another. Share at least one meal together each day, and use mealtimes to talk as a family. When parents heed that simple advice, their children enjoy greater well-being and do better at school.
Use mealtimes to talk as a family
In conclusion, ask yourself: What do I want out of life? What do I want for my family? If you desire a happier and more meaningful life, set priorities that reflect the proven wisdom found in the Bible.
IMPORTANT EPP MEMBERS
Thursday - October 5, 2017 @ 13:05:46
IMPORTANT EPP MEMBERS~
We need your help in our campaign to approve Scenesse
If you or someone you know is in the EPP MEDICAL
community (Doctor, RN, or Other Medical Professional) and
PORPHYRIA POST: Scenesse, FDA, and an EPP campaign updateâ?¦
Wednesday - September 20, 2017 @ 07:00:07
PORPHYRIA POST: Scenesse, FDA, and an EPP campaign updateâ?¦
CLINUVEL PROVIDES UPDATE ON SCENESSE FDA FILING
A company announcement from Clinuvel Pharmaceuticals was released on September 19, 2017 regarding their submission for approval to the FDA for the orphan drugSCENESSEwhich is used to treat Erythropoietic Protoporphyria. The announcement discusses both regulatory status and post-marketing safety data.
Clinuvel is seeking Priority Review for this product. This is a regulatory status that would decrease the review timeframe from the traditional 10 months to 6 months. The FDA response to this request is expected at the start of the review period.
Clinuvel closely monitors the safety profile of SCENESSEunder a European Post Authorization Safety Study (PASS) protocol. Clinuvel has agreed with the FDA that safety data from PASS will form part of the US post-marketing submission.
Dr. Scott Gottlieb, FDA Commissioner, spoke to the National Academy of Sciences yesterday on the impact of real world evidence on medical product development. He outlined opportunities for the FDA to include real world evidence in product development and regulatory decision-making. This is a positive sign that the FDA is looking outside of clinical data into the experience and voice of a patient.
During todays workshop, and in the years ahead, I want you to know that FDA will support your work on these efforts. At FDA, we intend to expand our regulatory policy development work on achieving the more appropriate adoption of RWE (real world evidence) as part of the entire life cycle of medical products. We cant do it alone. Your collaboration on these efforts is going to be critical to our success.
The APF will be launching another multi-faceted campaign seeking Scenesse approval. Stay tuned for further information and action alerts! It will take us all to participate to make this life-altering treatment available for ALL who seek it!
PATIENT MEETING WASHINGTON, DC
Members in the Washington, DC area are invited to attend a Patient Education and Support Meeting at the home of Kristen Wheeden, the APF Director of Development in Bethesda, MD. There will be a presentation and question/answer session with an on-site Porphyria expert. Come meet old and new friends who share your experiences with Porphyria. Please RSVP to Edrin Williams in the APF office at 1.866.APF.3635 or edrinw@porphyriafoundation.org. We look forward to seeing you!
You are invited to an American Porphyria Foundation Patient Education and Support Meeting
Sunday Oct 22, 2017 6:30PM-8:30PM EST
Question and Answer Session with a Porphyria Expert
Meet Old and New Friends who Share Tour Experiences with Porphyria
View the Latest Educational Material from the American Porphyria Foundation
At the Home of Kristen Wheeden
APF Director of Development
9007 Ewing Drive
Bethesda, MD 20817
Family and Friends are Welcome to Attend!
Please RSVP: 1.866.APF,3635 or Email: edrinw@porphyriafoundation.org
The biology of emotionand what it may teach us about helping people to live longer
Could a sunny outlook mean fewer colds and less heart disease?
Do hope and curiosity somehow protect against hypertension, diabetes, and respiratory tract infections?
Do happier people live longerand, if so, why?
These are the kinds of questions that researchers are asking as they explore a newand sometimes controversialavenue of public health: documenting and understanding the link between positive emotions and good health.
A vast scientific literature has detailed how negative emotions harm the body. Serious, sustained stress or fear can alter biological systems in a way that, over time, adds up to wear and tear and, eventually, illnesses such as heart disease, stroke, and diabetes. Chronic anger and anxiety can disrupt cardiac function by changing the hearts electrical stability, hastening atherosclerosis, and increasing systemic inflammation.
Jack P. Shonkoff, Julius B. Richmond FAMRI Professor of Child Health and Development at HSPH and at the Harvard Graduate School of Education, and Professor of Pediatrics at Harvard Medical School, explains that early childhood toxic stressthe sustained activation of the bodys stress response system resulting from such early life experiences as chronic neglect, exposure to violence, or living alone with a parent suffering severe mental illnesshas harmful effects on the brain and other organ systems. Among these effects is a hair-trigger physiological response to stress, which can lead to a faster heart rate, higher blood pressure, and a jump in stress hormones.
Focusing on the positive
But negative emotions are only one-half of the equation, says Laura Kubzansky, HSPH associate professor of society, human development, and health. It looks like there is a benefit of positive mental health that goes beyond the fact that youre not depressed. What that is is still a mystery. But when we understand the set of processes involved, we will have much more insight into how health works.
Kubzansky is at the forefront of such research. In a 2007 study that followed more than 6,000 men and women aged 25 to 74 for 20 years, for example, she found that emotional vitalitya sense of enthusiasm, of hopefulness, of engagement in life, and the ability to face lifes stresses with emotional balanceappears to reduce the risk of coronary heart disease. The protective effect was distinct and measurable, even when taking into account such wholesome behaviors as not smoking and regular exercise.
Keys to a happier, healthier life
Research suggests that certain personal attributeswhether inborn or shaped by positive life circumstanceshelp s
behaviors such as unsafe sex, drinking alcohol to excess, and regular overeating
Among dozens of published papers, Kubzansky has shown that children who are able to stay focused on a task and have a more positive outlook at age 7 report better general health and fewer illnesses 30 years later. She has found that optimism cuts the risk of coronary heart disease by half.
Kubzanskys methods illustrate the creativity needed to do research at the novel intersection of experimental psychology and public health. In the emotional vitality study, for example, she used information that had originally been collected in the massive National Health and Nutrition Examination Survey, or NHANES, an ongoing program that assesses the health and nutritional status of adults and children in the United States. Starting with the NHANES measure known as the General Well-Being Schedule, Kubzansky crafted an adaptation that instead reflected emotional vitality, and then scientifically validated her new measure. Her research has also drawn on preexisting data from the Veterans Administration Normative Aging Study, the National Collaborative Perinatal Project, and other decades-long prospective studies.
In essence, Kubzansky is leveraging gold-standard epidemiological methods to ask new public health questions. Im being opportunistic, she says. I dont want to wait 30 years for an answer.
Laura Kubzansky doesnt want her research on positive emotions to be used to blame people for getting sick.
State of mind=state of body
Some public health professionals contend that the apparent beneficial effects of positive emotions do not stem from anything intrinsically protective in upbeat mind states, but rather from the fact that positive emotions mark the absence of negative moods and self-destructive habits. Kubzansky and others disagree. They believe that there is more to the phenomenonand that scientists are only beginning to glean the possible biological, behavioral, and cognitive mechanisms.
Previous work supports this contention. In 1979, Lisa Berkman, director of the Harvard Center for Population and Development Studies, co-authored a seminal study of nearly 7,000 adults in Alameda County, California. Participants who reported fewer social ties at the beginning of the survey were more than twice as likely to die over the nine-year follow-up period, an effect unrelated to behaviors such as smoking, drinking, and physical activity. Social ties included marriage, contact with friends and relatives, organizational and church membership.
A happiness policy?
If scientists proved unequivocally that positive moods improve health, would policymakers act? Some observe that, in the U.S., we define happiness in economic termsthe pursuit of material goods. They contend that even an avalanche of research showing that emotional well-being protected health would have no traction in the policy world. Many Americans believe, after all, that people are responsible for their own lives.
But others see direct policy implications. In public health, its important to understand how we can translate guidelines into behavior, notes Eric Rimm, HSPH associate professor in the Departments of Epidemiology and Nutrition and director of the program in cardiovascular epidemiology. Seventy to 80 percent of heart attacks in this country occur not because of genetics nor through some mysterious causative factors. Its through lifestyle choices people make: diet, smoking, exercise. Why are people choosing to do these things? Does mood come into play?
The toll of toxic stress goes far beyond poorer health for individualspopulation-wide, the cost of chronic diseases related to these conditions is enormous. Imagine if we could enact a policy that would reduce heart disease by just 1 percent, suggests Shonkoff. How many billions of dollars and how many lives would that save? Now what if we could also reduce diabeteswhich is growing in epidemic proportionsand even stroke? The point, Shonkoff says, is that society pays a considerable cost for treating chronic diseases in adulthood, and reducing toxic stress early in life may actually get out in front of these diseases to prevent them.
A stress test of a different sort
In Laura Kubzanskys Society and Health Psychophysiology Labmodest and neutral as the blandest therapy officev
Kubzansky concedes that psychological states such as anxiety or depressionor happiness and optimismare forged by both nature and nurture. They are 4050 percent heritable, which means you may be born with the genetic predisposition. But this also suggests there is a lot of room to maneuver. Her dream prevention: instill emotional and social competence in childrenwith the help of parents, teachers, pediatricians, sports coaches, school counselors, mental health professionals, and policy makersthat would help confer not only good mental health but also physical resilience for a lifetime.
Even in adulthood, its not too late to cultivate these qualities, she says. While psychotherapy or meditation may work for one person, someone else may prefer faith-based activities, sports, or simply spending time with friends. My guess is that many of the people who are chronically distressed never figured out how to come back from a bad experience, focus on something different, or change their perspective.
Mapping happiness
Drawing on recently compiled data from a nationally representative study of older adults, Kubzansky is beginning to map what she calls the social distribution of well-being. She is working with information collected on participants sense of meaning and purpose, life satisfaction, and positive mood. By tracking how these measures and health fall out across traditional demographic categories such as race and ethnicity, education, income, gender, and other categories, she hopes to understand in a fine-grained way what it is about certain social environments that confers better frame of mind and better physical health.
The last thing she wants, Kubzansky says, is for her research to be used to blame people for not simply being happierand therefore healthier. Referring to one of her first major studies, which found a link between worry and heart disease, she said: My biggest fear was that journalists would pick it up and the headlines would be, Dont worry, be happy. Thats useless. Not everyone lives in an environment where you can turn off worry. When you take this research out of the social context, it has the potential to be a slippery slope for victim blaming.
Being in the moment
Kubzansky, who is married and has two young children, says her work has made her think a lot more about finding balance in her own life. To that end, she says, she recently signed up for a yoga class. She also plays classical pianoboth chamber music with friends and solo hours at the keyboard for her own enjoyment.
When Im playing piano, she explains, Im in the moment. Im not worrying or thinking or trying to work out a problem. Im just doing this thing that takes all my attention.
That insight is also at the center of her research. Everyone needs to find a way to be in the moment, she says, to find a restorative state that allows them to put down their burdens.
Calling: Research Research Research!
Thursday - September 14, 2017 @ 07:00:23
Research Volunteers Needed
RESEARCH: YOU ARE NEEDED!
We are seeking patients to join our research studies. Get involved today!
Longitudinal Study:
Participation in this study is very easy. It does not require you to travel to the research site. The study involves a review of your medical records, completing comprehensive questionnaires about your symptoms and quality of life, and providing blood and urine samples. You will be working closely with porphyria experts and researchers.
The purpose of this long-term follow-up study is to provide a better understanding of the natural history of porphyrias, as affected by available therapies, and to aid in developing new forms of treatment.
All Porphyrias
Panhematin Study:
This clinical trial compares Panhematin to glucose to determine the effectiveness of Panhematin as treatments for acute attacks.
AIP, VP & HCP
Contact Edrin at the APF for additional information.
Harvoni Study for PCT:
This clinical trial will assess whether Harvoni alone is an effective and durable treatment for PCT in patients with both HCV and PCT. This trial will also determine whether treating patients with PCT and HCV with Harvoni is as effective as treating PCT with standard therapies, phlebotomy or hyrdoxychloroquine.
Therefore, the purpose of this study is to determine if giving a standard treatment for HCV is also an effective treatment for PCT.
Contact Edrin at the APF for additional information.
Iron Therapy for EPP:
This research study aims to determine if oral iron can decrease erythrocyte protoporphyrin levels in patients with EPP or XLP, to assess whether oral iron can improve iron status and decrease plasma porphyrin levels and symptoms in patients with EPP or XLP and assess if there is an improvement in the quality of life in treated patients.
Therefore, the purpose of this study is to determine the effect of oral iron in EPP and XLP patients.
In order to participate in a study, you must personally contact the study coordinator of the participating institution closest to you by phone or e-mail. Please contact Edrin at the APF at 1.866.APF.3635 or 713.266.9617 for further information.
Porphyria Cutanea Tarda What it is! Know the facts!
Thursday - September 14, 2017 @ 07:00:16
Porphyria Cutanea Tarda
Important
It is possible that the main title of the report Porphyria Cutanea Tarda is not the name you expected. Please check the synonyms listing to find the alternate name(s) and disorder subdivision(s) covered by this report.
Porphyria cutanea tarda (PCT) is a type of porphyria in which affected individuals are sensitive to sunlight. Exposed skin shows abnormalities that range from slight fragility of the skin to persistent scarring and disfiguration. Due to fragility of the skin, minor trauma may induce blister formation. Areas of increased and decreased pigment content may be noted on the skin. Blistering of light exposed skin and increased hair growth are also characteristic.
PCT is caused by a deficiency of the uroporphyrinogen decarboxylase (URO-D) enzyme in the liver. The disorder can be acquired or can be caused by an inherited gene mutation in the UROD gene. The inherited form of PCT is also called familial PCT and follows autosomal dominant inheritance. Many individuals with a UROD gene mutation never experience symptoms of the disease.
PCT becomes active and causes symptoms when triggered by an environmental factor that affects liver cells (hepatocytes). These environmental factors include alcohol, estrogens, hepatitis C, and human immunodeficiency viruses (HIV). Individuals who have disorders that lead to excess iron in tissues such as hemochromatosis also have an increased risk of developing PCT. It is usually necessary for an environmental trigger to be present to cause symptoms of either the acquired or inherited type of PCT.
Resources
CLIMB (Children Living with Inherited Metabolic Diseases) Climb Building 176 Nantwich Road Crewe, CW2 6BG United Kingdom Tel: 4408452412173 Fax: 4408452412174 Email: enquiries@climb.org.uk Internet: http://www.CLIMB.org.uk
American Porphyria Foundation 4900 Woodway, Suite 780 Houston, TX 77056-1837 Tel: (713)266-9617 Fax: (713)840-9552 Tel: (866)273-3635 Email: porphyrus@aol.com Internet: http://www.porphyriafoundation.com
NIH/National Institute of Diabetes, Digestive & Kidney Diseases Office of Communications & Public Liaison Bldg 31, Rm 9A06 31 Center Drive, MSC 2560 Bethesda, MD 20892-2560 Tel: (301)496-3583 Email: NDDIC@info.niddk.nih.gov Internet: http://www2.niddk.nih.gov/
MedicAlert Foundation International 2323 Colorado Avenue Turlock, CA 95382 USA Tel: (209)669-2401 Fax: (209)669-2456 Tel: (888)633-4298 Email: Inquiries@medicalert.org Internet: http://www.medicalert.org
Genetic and Rare Diseases (GARD) Information Center PO Box 8126 Gaithersburg, MD 20898-8126 Tel: (301)251-4925 Fax: (301)251-4911 Tel: (888)205-2311 TDD: (888)205-3223 Internet: http://rarediseases.info.nih.gov/GARD/
Read the Experience of Kathryn Nelson & PCT
Monday - September 11, 2017 @ 07:00:18
My Porphyria Cutanea Tarda (PCT) experience began roughly seven years ago. At the time I was living in Irving, Texas. When lesions appeared on my face, forearms and legs, I thought that perhaps it was related to Psoriasis, an autoimmune disorder I had since I was a child. As a result, I resorted to a fairly common approach to Psoriasis which is exposure to UV rays. I spent an hour or so every afternoon in the sun, but more lesions developed and the existing ones grew in size. Repeated visits to my Dermatologist resulted in a variety of diagnoses including eczema, hives and finally a "picking" disorder which basically meant the doctor believes you are picking at your skin causing sores and infections. His primary reason for this was that the sores appeared only where I could physically reach the area, in other words, there were no lesions on my back or the backs of my legs. I pointed out that as a forty plus year old woman, I didn't think I had suddenly started to pick my skin but he was not deterred.
I continued to use antibiotic ointments on the most severe patches and, as a result of other health concerns, discontinued my efforts to get a daily dose of UV rays. Over time, the lesions began to shrink but a new symptom developed. My skin darkened dramatically in areas where there were no blisters and where the blisters had healed, the skin turned white, giving the appearance of vitiligo. I was seeing a Rheumatologist for other autoimmune symptoms and he suggested I visit a dermatologist he knew who specialized in autoimmune skin disorders. He also prescribed Plaquenel to treat lupus-like symptoms tied to an undifferentiated connective tissue disorder.
It was several months later when I met with Dr. Melissa Costner in Dallas. By that time, most of the lesions and blisters were healed, however, the splotchy dark patches and scarred white areas covered most of my arms, face and large portions of my legs. I described my experiences over the previous three years. Dr. Costner listened closely and then said she wanted to do some blood work. Three weeks later, I returned to her office where she explained I had a familial type of PCT. In one minute, all of the heartaches of the previous four years made sense. She listened to me, something the previous doctors failed to do. She believed me when I said I wasn't "picking" my skin and said that rather than affecting areas where I "could reach," PCT was actually affecting areas being exposed to the sun. By reducing the time I was spending in the sun due to my energy issues, I had actually started the PCT healing process.
Today I still have very dark patches on my skin and very white scars but I have few reoccurrences of PCT. I play close attention to the medications I take, particularly since I have other auto-immune disorders which require a heavy regimen of treatment. PCT was not something I had ever heard of and was honestly not my biggest health concern at the time. But finding a doctor who listened gave me a sense of empowerment that I continue rely on today in all aspects of my health and with all my doctors.
EPP Informative Read from New Zeeland
Friday - September 8, 2017 @ 07:00:07
Erythropoietic protoporphyria pathology
Author: Dr Harriet Cheng, Dermatology Registrar, Waikato Hospital, Hamilton, New Zealand, 2013.
Erythropoietic protoporphyria (EPP) is a metabolic condition caused by deficiency of the ferrochelatase enzyme leading to intracellular accumulation of haeme precursors. Presentation is with acute photosensitivity in childhood giving rise to erythema and scale and eventually scarring and wax-like thickening of the skin.
Histology of erythropoietic protoporphyria
In the dermiseosinophilic hyaline material is deposited in and around blood vessel walls. The deposition can be extensive and involve the surrounding dermis to mimic a colloid milium (figures 1,2). Impressive thickening of vessels is seen at higher power examination (figure 3).
Subepidermal blisters similar to those seen in porphyria cutanea tarda are seen in early lesions of EPP. These blisters are cell-poor and inflammatoryinfiltrate is sparse. Caterpillar bodies (linear segmented structures composed of degenerating keratinocytes and basement membrane components) may be seen in the basal layerepidermisoverlying blisters.
Erythropoietic protoporphyriapathology
Figure 1
The hyaline material surrounding blood vessels is PAS-positive (figure 4) and diastase-resistant. Direct immunofluorescence shows deposition of immunoglobulin (especially IgG), fibrin, and C3 around blood vessels in the papillary dermis. Deposition of the membrane attack complex (C5-9) has also been demonstrated in vessel walls.The material also stains positively with Sudan black and Hales colloidal iron method. Caterpillar bodies are PAS-positive.
Differential diagnosis of erythropoietic protoporphyria
Porphyria cutanea tarda: Histological appearances are very similar. Typically vascular depositis in porphyria cutanea tarda are less pronounced than EPP and limited to the vessel wall. Solar elastosis (often present in PCT due to patient age) is not a feature of EPP.
Scleroderma: The dermal changes of chronic EPP may resemble scleroderma; however, the collagen in EPP is thought to have a looser texture. Blistering and vascular cuffing are not features of scleroderma.
Lipoid proteinosis: Vascular cuffing is a feature of both conditions; however, in EPP, cuffing does not extend around adnexal structures. Clinical correlation can be helpful.
Colloid milium: EPP closely resembles colloid milium when the deposition in the papillary dermis is extensive (figures 1, 2). In colloid milium, clefts form within the material and vascular thickening is not prominent.
Differentiation from other conditions with cell-poor subepidermal blistering (epidermolysis bullosa, pseudoporphyria and bullousamyloidosis) is on the basis of clinical features, immunofluorescence studies and serum, faecal and urine porphyrin testing.
Robin Davis Discusses EPP & Disabled Parking Permits
Wednesday - September 6, 2017 @ 07:00:12
Disabled Parking Permits: A Blessing for EPP Patients
I feel blessed that the Secretary of State in Michigan issued me a disabled parking permit. I know how important it can be to those of us who live with such a rare disorder, so I thought I'd share my story in hopes that it will assist others in obtaining a disabled permit. I also hope my story will open more eyes to the fact that EPP is in many ways a disability.
I've had Erythropoietic Protoporphyria (EPP) symptoms since I was 10 months old. When I was very young I was teased and emotionally tormented by the kids in school due to my EPP. They would call me named like Fish, Scales, Scabs or any other name they could think of till I'd break down crying or run home. They didn't understand what EPP was, they only saw that my face and arms were swollen with rashes, and itching until my skin was raw. It was hard to take while I was growing up, but then as I got older I realized children are not always nice. What's surprising is some of those children seem to never grow up and still get some kind of pleasure out of making fun of others or of what they do not understand.
Back in 1992, my doctor told me he wanted me to apply for a handicap parking permit, not wanting me exposed to the sun any more than absolutely necessary. My doctor wrote a letter to the Secretary of State's office explaining what EPP is, the danger of sun exposure and how quickly the sun can affect a person with EPP. When I took the letter to make my application for a parking permit, I was shocked at the way I was treated.
The man who waited on me began to laugh as soon as he read the letter from my doctor. Then in a loud voice he said "we don't issue handicap permits because someone can't go in the sun, there are people out there with real medical problems, I suggest you put some clothes on." I couldn't understand his remark considering I was wearing a business suit at the time. I tried to explain the situation, speaking softly to make it clear I didn't want the entire room to hear our conversation. Rather than listening to me, this man just repeated what he had said, only louder. I walked out fighting my tears, shocked and embarrassed as everyone in the room stared at me.
My husband advised me to call the state directly to report my experience. I called and explained what had happened and how unprofessionally I was treated, and that fact that I have been tormented my entire life over this disorder by people who don't understand. The person I spoke to asked to me write a letter regarding my experience and to enclose the letter from my doctor and send it to the statewide Secretary of State's office. Within a week I had a letter of apology and my parking permit.
The letter from the Secretary of State's office mentioned that their department has guidelines at the local offices for how staff should deal with health issues. The department assured me that they would put a policy into effect instructing the staff to refer uncommon problems to the state-wide office for review. Applicants for disabled parking permits who have non-standard health problems will be notified by mail of the decision on their permits.
Best Wishes and God Bless.
Part 4 NIH Porphyria Basics (Updated Information)
Tuesday - September 5, 2017 @ 22:57:13
Part 4
Clinical Trials
The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and other components of the National Institutes of Health (NIH) conduct and support research into many diseases and conditions.
What are clinical trials, and are they right for you?
Clinical trials are part of clinical research and at the heart of all medical advances. Clinical trials look at new ways to prevent, detect, or treat disease. Researchers also use clinical trials to look at other aspects of care, such as improving the quality of life for people with chronic illnesses. Find out if clinical trials are right for you .
What clinical trials are open?
Clinical trials that are currently open and are recruiting can be viewed at www.ClinicalTrials.gov .
This content is provided as a service of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), part of the National Institutes of Health. The NIDDK translates and disseminates research findings through its clearinghouses and education programs to increase knowledge and understanding about health and disease among patients, health professionals, and the public. Content produced by the NIDDK is carefully reviewed by NIDDK scientists and other experts. The NIDDK would like to thank: Herbert L. Bonkovsky, M.D., Carolinas Health Care System
Part 3 NIH Porphyria Basics (Updated Information)
Monday - September 11, 2017 @ 14:51:35
Part 3
Points to Remember
Porphyrias are rare disorders that affect mainly the skin or nervous system and may cause abdominal pain.
Each of the eight types of porphyria corresponds to low levels of a specific enzyme in the heme biosynthetic pathway.
The exact rates of porphyria are unknown and vary around the world. For example, porphyria cutanea tarda is most common in the United States, and variegate porphyria is most common in South America.
Most porphyrias result from inheriting an abnormal gene, also called a gene mutation, from one parent.
Porphyria cutanea tarda is usually an acquired disorder, meaning factors other than genes cause the enzyme deficiency.
Symptoms of cutaneous porphyrias include
oversensitivity to sunlight
blisters on exposed areas of the skin
itching and swelling on exposed areas of the skin
Symptoms of acute porphyrias include
pain in the abdomen
pain in the chest, limbs, or back
nausea and vomiting
constipation
urinary retention
confusion
hallucinations
seizures and muscle weakness
A health care provider diagnoses porphyria with blood, urine, and stool tests.
Treatment for porphyria depends on the type of porphyria the person has and the severity of the symptoms.
References
APF Office Open 9/6/17
Friday - September 1, 2017 @ 07:00:00
The American Porphyria Foundation will resume normal business operations beginning Tuesday, September 6, 2017. We appreciate your thoughts, prayers, patience and understanding during this time. The APF staff remained safe during this storm and will be diligently working to get to your request by responding to your calls and emails.
"Remember.Research is the key to your cure!"
Part 2 NIH 101 On Porphyrias (Updated Information)
Wednesday - August 30, 2017 @ 14:00:16
Part 2
Eating, Diet, and Nutrition
People with an acute porphyria should eat a diet with an average-to-high level of carbohydrates. The recommended dietary allowance for carbohydrates is 130 g per day for adults and children 1 year of age or older; pregnant and breastfeeding women need higher intakes.2 People should avoid limiting intake of carbohydrates and calories, even for short periods of time, as this type of dieting or fasting can trigger symptoms. People with an acute porphyria who want to lose weight should talk with their health care providers about diets they can follow to lose weight gradually. People undergoing therapeutic phlebotomies should drink plenty of milk, water, or juice before and after each procedure. A health care provider may recommend vitamin and mineral supplements for people with a cutaneous porphyria.
porphyriafoundation.org
APF is #Houston Strong!
Wednesday - August 30, 2017 @ 07:00:39
The American Porphyria Foundation office located in Houston, TX remains closed due to the extreme flooding from Hurricane Harvey. All office staff are safe. Please continue to contact us with any needs as we will be checking voicemail and email on a regular basis. We will inform you when the office has re-opened. For those with questions that need to be answered with urgency please contact Desiree Lyon, Executive Director at 713.857.0995. Thank you.
WOODWAY DRIVE - Directly across the street from where the APF office is located.
NIH Part 1 101 on Porphyrias (Updated Info)
Friday - August 25, 2017 @ 07:00:25
Part 1
Porphyria
What are porphyrias?
Porphyrias are rare disorders that affect mainly the skin or nervous system and may cause abdominal pain. These disorders are usually inherited, meaning they are caused by abnormalities in genes passed from parents to children. When a person has a porphyria, cells fail to change body chemicals called porphyrins and porphyrin precursors into heme, the substance that gives blood its red color. The body makes heme mainly in the bone marrow and liver. Bone marrow is the soft, spongelike tissue inside the bones; it makes stem cells that develop into one of the three types of blood cellsred blood cells, white blood cells, and platelets. The process of making heme is called the heme biosynthetic pathway. One of eight enzymes controls each step of the process. The body has a problem making heme if any one of the enzymes is at a low level, also called a deficiency. Porphyrins and porphyrin precursors of heme then build up in the body and cause illness.
What is heme and what does it do?
Heme is a red pigment composed of iron linked to a chemical called protoporphyrin. Heme has important functions in the body. The largest amounts of heme are in the form of hemoglobin, found in red blood cells and bone marrow. Hemoglobin carries oxygen from the lungs to all parts of the body. In the liver, heme is a component of proteins that break down hormones, medications, and other chemicals and keep liver cells functioning normally. Heme is an important part of nearly every cell in the body.
What are the types of porphyria?
Each of the eight types of porphyria corresponds to low levels of a specific enzyme in the heme biosynthetic pathway. Experts often classify porphyrias as acute or cutaneous based on the symptoms a person experiences:
Acute porphyrias affect the nervous system. They occur rapidly and last only a short time.
Cutaneous porphyrias affect the skin.
Two types of acute porphyrias, hereditary coproporphyria and variegate porphyria, can also have cutaneous symptoms. Experts also classify porphyrias as erythropoietic or hepatic:
In erythropoietic porphyrias, the body overproduces porphyrins, mainly in the bone marrow.
In hepatic porphyrias, the body overproduces porphyrins and porphyrin precursors, mainly in the liver.
Table 1 lists each type of porphyria, the deficient enzyme responsible for the disorder, and the main location of porphyrin buildup. Table 1. Types of porphyria
The exact rates of porphyria are unknown and vary around the world. For example, porphyria cutanea tarda is most common in the United States, and variegate porphyria is most common in South America.1
What causes porphyria?
Most porphyrias are inherited disorders. Scientists have identified genes for all eight enzymes in the heme biosynthetic pathway. Most porphyrias result from inheriting an abnormal gene, also called a gene mutation, from one parent. Some porphyrias, such as congenital erythropoietic porphyria, hepatoerythropoietic porphyria, and erythropoietic protoporphyria, occur when a person inherits two abnormal genes, one from each parent. The likeliness of a person passing the abnormal gene or genes to the next generation depends on the type of porphyria. Porphyria cutanea tarda is usually an acquired disorder, meaning factors other than genes cause the enzyme deficiency. This type of porphyria can be triggered by
too much iron
use of alcohol or estrogen
smoking
chronic hepatitis Ca long-lasting liver disease that causes inflammation, or swelling, of the liver
HIVthe virus that causes AIDS
abnormal genes associated with hemochromatosisthe most common form of iron overload disease, which causes the body to absorb too much iron
For all types of porphyria, symptoms can be triggered by
use of alcohol
smoking
use of certain medications or hormones
exposure to sunlight
stress
dieting and fasting
What are the symptoms of porphyria?
Some people with porphyria-causing gene mutations have latent porphyria, meaning they have no symptoms of the disorder. Symptoms of cutaneous porphyrias include
oversensitivity to sunlight
blisters on exposed areas of the skin
itching and swelling on exposed areas of the skin
Symptoms of acute porphyrias include
pain in the abdomenthe area between the chest and hips
pain in the chest, limbs, or back
nausea and vomiting
constipationa condition in which an adult has fewer than three bowel movements a week or a child has fewer than two bowel movements a week, depending on the person
urinary retentionthe inability to empty the bladder completely
confusion
hallucinations
seizures and muscle weakness
Symptoms of acute porphyrias can develop over hours or days and last for days or weeks. These symptoms can come and go over time, while symptoms of cutaneous porphyrias tend to be more continuous. Porphyria symptoms can vary widely in severity.
How is porphyria diagnosed?
A health care provider diagnoses porphyria with blood, urine, and stool tests. These tests take place at a health care providers office or a commercial facility. A blood test involves drawing blood and sending the sample to a lab for analysis. For urine and stool tests, the patient collects a sample of urine or stool in a special container. A health care provider tests the samples in the office or sends them to a lab for analysis. High levels of porphyrins or porphyrin precursors in blood, urine, or stool indicate porphyria. A health care provider may also recommend DNA testing of a blood sample to look for known gene mutations that cause porphyrias.
How is porphyria treated?
Treatment for porphyria depends on the type of porphyria the person has and the severity of the symptoms.
Acute Porphyrias
A health care provider treats acute porphyrias with heme or glucose loading to decrease the livers production of porphyrins and porphyrin precursors. A patient receives heme intravenously once a day for 4 days. Glucose loading involves giving a patient a glucose solution by mouth or intravenously. Heme is usually more effective and is the treatment of choice unless symptoms are mild. In rare instances, if symptoms are severe, a health care provider will recommend liver transplantation to treat acute porphyria. In liver transplantation, a surgeon removes a diseased or an injured liver and replaces it with a healthy, whole liver or a segment of a liver from another person, called a donor. A patient has liver transplantation surgery in a hospital under general anesthesia. Liver transplantation can cure liver failure. More information is provided in the NIDDK health topic, Liver Transplantation.
Cutaneous Porphyrias
The most important step a person can take to treat a cutaneous porphyria is to avoid sunlight as much as possible. Other cutaneous porphyrias are treated as follows:
Porphyria cutanea tarda. A health care provider treats porphyria cutanea tarda by removing factors that tend to activate the disease and by performing repeated therapeutic phlebotomies to reduce iron in the liver. Therapeutic phlebotomy is the removal of about a pint of blood from a vein in the arm. A technician performs the procedure at a blood donation center, such as a hospital, clinic, or bloodmobile. A patient does not require anesthesia. Another treatment approach is low-dose hydroxychloroquine tablets to reduce porphyrins in the liver.
Erythropoietic protoporphyria. People with erythropoietic protoporphyria may be given beta-carotene or cysteine to improve sunlight tolerance, though these medications do not lower porphyrin levels. Experts recommend hepatitis A and B vaccines and avoiding alcohol to prevent protoporphyric liver failure. A health care provider may use liver transplantation or a combination of medications to treat people who develop liver failure. Unfortunately, liver transplantation does not correct the primary defect, which is the continuous overproduction of protoporphyria by bone marrow. Successful bone marrow transplantations may successfully cure erythropoietic protoporphyria. A health care provider only considers bone marrow transplantation if the disease is severe and leading to secondary liver disease.
Congenital erythropoietic porphyria and hepatoerythropoietic porphyria. People with congenital erythropoietic porphyria or hepatoerythropoietic porphyria may need surgery to remove the spleen or blood transfusions to treat anemia. A surgeon removes the spleen in a hospital, and a patient receives general anesthesia. With a blood transfusion, a patient receives blood through an intravenous (IV) line inserted into a vein. A technician performs the procedure at a blood donation center, and a patient does not need anesthesia.
Secondary Porphyrinurias
Conditions called secondary porphyrinurias, such as disorders of the liver and bone marrow, as well as a number of drugs, chemicals, and toxins are often mistaken for porphyria because they lead to mild or moderate increases in porphyrin levels in the urine. Only highnot mild or moderatelevels of porphyrin or porphyrin precursors lead to a diagnosis of porphyria.
RNA interference (RNAi) specialist Alnylam has hit the accelerator on its candidate drug for ultrarare porphyria diseases, prepping for phase 3 trials later this year after unveiling initial results in 12 patients.
Alnylam's givosiran achieved a dramatic reduction in annual attack rates in the phase 1 trial reported at the International Congress on Porphyrins and Porphyrias in Bordeaux, France, and could become the first drug to be approved for preventing attacks, according to the biotech.
It's a needed boon for Alnylam, which was hit hard last year after it was forced to drop its hereditary ATTR amyloidosis drug revusiran. More patient deaths were seen on that drug versus placebo in the late-stage ENDEAVOUR trial, forcing a switch to new lead drug patisiran.
Porphyrias are a group of disorders that result from a build-up of substancescalled porphyrinsthat can damage organs and the nervous system. As the name suggests, acute hepatic porphyrias damage the liver, and are among the most common forms of the disease, but are still very uncommon. Taken together, all forms of porphyria afflict fewer than 200,000 people in the U.S., according to the American Porphyria Foundation (APP).
The most common symptom is intense abdominal pain, but some people also suffer muscular weakness, sensory disturbances or convulsions, and as a group the diseases are "devastating for patients" with attacks lasting for days and often requiring admission to hospital, according to Akshay Vaishnaw M.D., Ph.D., Alnylam's head of R&D, who discussed the data on a conference call this morning.
At the moment, the only drug approved for use in porphyria is hemin, which is labeled as a treatment for attacks but is given off-label to some patients for prevention. Intravenous glucose therapy is also used, but both treatments have limited activity in preventing or hastening recovery from attacks, according to the APP.
Enter givosiran, a gene-silencing drug designed to switch off the aminolevulinic acid synthase 1 (ALAS1) gene. The drug reduces levels of two porphyrin precursors (ALA and PBG), which researchers hope will prevent attacks.
The RNAi switched off ALAS1 in the trial and achieved a 73% reduction (PDF) in annualized attack rate compared to placebo in the 12-patient phase 1 trial, with a 73% reduction in annualized hemin doses. Clinicians have suggested a 30% reduction in attacks would be clinically meaningful, according to Barry Greene, president of Alnylam.
Importantly, the reduction in attacks appeared to be dose-dependent and matched reductions in ALA/PBG levels, suggesting these could be used as biomarkers for the drug's activity in future studies, said Vaishnaw. There was one death among patients in the treatment group (due to hemorrhagic pancreatitis) as well as three other serious adverse events that the investigators concluded were not linked to the study drug.
Data from an eight-patient open-label extension study found a further reduction in attacks and suggested control could be better with longer-term dosing, said Vaishnaw, although he stressed the results are still preliminary with such a small patient cohort. That said, there is no evidence for this with hemin, but some data suggest that control gets worse in hemin-treated patients over time, he added.
Either way, "there is no question these annualized attack rate reductions have a big impact on the lives of patients," said Vaishnaw on the conference call.
"We are very encouraged by the data â?¦ and the potential for the givosiran program," said Greene, noting that the drug could offer a once-monthly, low-volume subcutaneous injection therapy for patients with porphyrias.
On the thorny topic of pricing, Alnylam is thinking ahead and already reckons it can make a big impact on the costs of acute hepatic porphyrias. Treatment currently costs between $400,000 and $650,000 per patient per year, according to a natural history study (EXPLORE) due for presentation at the ICPP meeting on Wednesday
The phase 3 program will get underway in the fourth quarter and focus on attack ratesthe most devastating element of the diseasesaid Alnylam. It will, however, also include a pharmacoeconomic analysis to take into account direct healthcare costs as well as indirect factors such as loss of work days, etc.
Alnylamwhich said it plans to self-develop and commercialize the drughas already secured breakthrough registration for the drug in the U.S. and PRIME (priority medicine) status in the E.U., so if the phase 3 program goes well, givosiran is in line for speedy review by regulators.
A Little Bit Of History of Porphyria
Monday - August 21, 2017 @ 07:00:31
Medical Moment~ History of Porphyria A Little Bit of History 1841 - The term porphyrin comes from the Greek word, porphyus, meaning reddish-purple. It was first thought that the reddish color of blood was from iron. One early scientist performed an experiment to prove that this was not the case. He washed dried blood with concentrated sulfuric acid to free the iron. He then treated it with alcohol and the resulting iron free residue took on a reddish purple color though it contained no iron compound 1844 - Gerardus Johannes Mulder determined the chemical composition of this purplish, iron free substance, which he named "hematin," He also illustrated that hematin took up oxygen. 1867 - J.L.W. Thudichum described the beautiful spectrum and fluorescence of these red porphyrins after he published his first book on the analysis of urine. 1871 - Felix Hoppe-Seyler crystallized hematin and described its spectrum. He then demonstrated that the crystalline form differed from one animal species to another. Using his own newly constructed gas pump, he found that oxygen formed a loose, dissociable compound with hemoglobin, which he called "oxyhemoglobin." He renamed the iron free hematin hematoPorphyrin. 1874 - Dr. J.H. Schultz described a case of a 33-year-old male weaver who suffered from skin sensitivity, an enlarged spleen and reddish urine since he was an infant. He called the condition pempigus leprosus. His was most likely the first description of protoporphyria. Dr. Schultz was later credited with giving the disease its name. 1880 - MacMunn described a patients dark reddish urine of a patient with symptoms of an attack of acute Porphyria. 1888 - Shortly after, sulphonal was introduced as a hypnotic drug, Joseph Stokvis had a patient who, after taking the drug, excreated the tell-tale dark reddish urine typical of porphyria. The elderly woman then became paralyzed and died. Stokvis deducted that the pigment in her urine was the hematoporphyrin. 1889 - B.J.Stokvis published the first case and clinical description of acute hepatic porphyria. 1890 - George Harley (1829-96) studied a 27-year-old who also excreted reddish urine and an "unusual nerve disturbance after taking sulphonal. 1898 - T.McCall Anderson described two brothers had eruptions with burning and pruitus on the sun exposed areas of their skin so severe that they lost part of their ears and nose. They exhibited dark urine. 1898 - Alfred F. Harris demonstrated that the urine of both brothers contained the hematoporphyrin group. 1906 - Dr. Max Dobrschansky described the first case of acute porphyria after a patient had a barbiturate. 1911 - H. Gunther classified the diseases of porphyria, including congenital erythropoietic porphyria (CEP), which he called congenital hepatoporphyria, the rarest porphyria. 1913 - Dr. Friedrich Meyer Betz injected himself with hematoporphyrins to determine their photodynamic impact. He subjected himself to the sun and became so photosensitized that the extremely painful photosensitive effect lasted several months. The photos of Dr. Betz taken hours after he injected himself illustrated his badly swollen face. He was unrecognizable until the swelling decreased. The-experiment is used today in dermatology text books. View these photos on the APF website. 1915 - Hans Fischer studied one of H. Gunthers patients, Mr. Petry, who had the rare type of Porphyria, CEP. Using data from Mr. Petrys case, Fischer provided significant insight into the chemistry of porphyrins. He also found that uroporphyrins and coproporphyrins were different from hematoporphyrins and subsequently suggested that the hemato prefix be dropped. 1923 - A. E. Garrod credits H. Gunther with first recognizing that hematoporphyria was, in fact, an inherited metabolic problem in his manuscript, Inborn Errors of Metabolism. This is the first time the term "inborn errors" of metabolism had been ever used for a group of inherited metabolic disorders and the year CEP was first identified. 1937 Dr. Jan G. Waldenstrom suggested that the name of the diseases of porphyrin metablolism be porphyrias rather than Hematoporphyrias. Using Paul Ehrlichs aldehyde reagent, Waldenstorm identified 103 patients with acute porphyria by testing their urine and noting the red color. He discovered that asymptomatic family members of these patients also had the same reaction if they ingested even small amounts of barbiturates and sulphonal. 1949 -Dr. Cecil J. Watson identified cases in which there were excessive amounts of coproporphyrins in the stool and urine and suggested that this was caused by an inborn error of metabolism. He continued his research in the United States, where he and Dr. Samuel Schwartz discovered a fundamental test, the "Watson-Schwartz tests". 1954 - R. Schmid, Samuel Schwartz and Cecil. J. Watson classified the porphyrias according to the porphyrin content in the bone marrow and liver. 1955 - A. Goldberg and H. Berger showed that individuals with an excess of coproporphyrin had another inherited form of porphyria that they called hereditary coproporphyria. HCP is an autosomal dominant form of hepatic porphyria that is very similar to acute intermittent porphyria, except that some patients develop skin photosensitivity, too. 1960's Earnest Porphyria research in Europe and US. 1961 - Heinrich Gustav Magnus described erythropoietic protoporphyria (EPP) as a genetic disorder arising from impaired activity of ferrochelatase, which is what adds iron to protoporphyrin to form heme. 1970-2011 - Drs. Anderson, Desnick, Bissell, Bloomer, Bonkovsky,, Bottomley, Dailey, Galbraith, ,Kappas, Kreimer-Birnbaum, Kushner, Lamon, Levere, Levine, Mathews-Roth, McDonaugh, Nichols, Peters, Sinclair, Pimstone, Pierach, Poh-Fitzpatrick, Sassa, Shedlofsky, Schmid, Sassa, Tishler, Tschudy, Watson,, Phillips and many others too numerous to name have furthered porphyria research and have bettered the health care of all of us with Porphyria. We owe all these people a great debt and a great measure of thanks. 2008-2011- The APF Protect the Future program to train the next generation of experts was initiated. We are grateful for the newest experts; Drs. Manisha Balwani, Lawrence Lui, Gagen Sood, Manish Thapar, Bradley Freilich, Charles Lourenco, Brenden McGuire, Bruce Wang, Majid Rizk, Guiherme Perini, Jennifer Guy, Jeffery Wickliffe, Aswani Singal, Sajid Mittal,Charles Parker.
EPP Have you ever wondered why you have been sunburnt on a cloudy day?
Thursday - August 17, 2017 @ 07:00:11
EPP
Have you ever wondered why you have been sunburnt on a cloudy day?
The total dose of UV radiation reaching the earths surface and hence, the potential damage to human skin and tissues, varies, depending on many factors.
The suns elevation in the sky depends on the time of the day and year. The shorter the distance that photons (making up the total of UV radiation) need to travel though the earth's atmosphere, the greater the intensity of UV radiation. The altitude of a location also effects UV radiation levels as the higher a location is above sea level, the shorter the distance UV radiation travels.
The thinning of the ozone layer located above Antarctica has had a considerable impact on the ability of the atmosphere to absorb UVB, a significant contributor to the increased incidence of skin cancer and other damage to human tissues which has been observed in populations bordering the ozone hole.
Clouds act on UV primarily by scattering radiation which can both reduce and enhance the UV radiation levels depending on the type of cloud cover.
Some clouds absorb infrared radiation and as a result of the diminished heat sensation, people are given a false sense of security and often change their behaviour on cloudy days, unaware that they are exposing themselves to this potential danger.
UV radiation is also reflected from surfaces such as sand, snow and water. These surfaces can increase the UV radiation at ground level and increase the amount of skin damage incurred from UV radiation exposure.
So, when all these factors are considered, it is important to recognise that the net potential UV risk is a result of these associated variables, depending an Individuals circumstances. The only sure way to significantly reduce the risk of skin damage is with vigilant protection from UV radiation and light, known as photoprotection.
EPP is CLINUVEL's lead clinical indication for SCENESSE(afamelanotide 16mg)
Monday - August 14, 2017 @ 15:15:57
EPP is CLINUVEL's lead clinical indication for SCENESSE(afamelanotide 16mg)
Clinical and regulatory progress:
In December 2014, the European Commission approved SCENESSE to prevent phototoxicity in adult patients diagnosed with EPP, following a recommendation from the European Medicines Agency's Committee for Medicinal Products for Human Use (CHMP). You can read the announcement here.
CLINUVEL is currently working to make SCENESSE available across Europe. If you have EPP and would like to receive updates on our program, please contact us.
EPP is a rare life-long genetic disease found mainly in fair-skinned people. It is characterised by severe phototoxicity (intolerance of light) of the skin resulting in intolerable pain, swelling and scarring, usually of exposed areas such as the face, hands and feet. Reactions can vary from mild to extreme with hospitalisation and powerful pain killers required in the worst cases.
Children and adults living with EPP must avoid sunlight and even reflected light for life, often staying indoors or wearing protective clothing. Conventional sunscreens have little to no effect.
Since sun avoidance is recommended, patients lead lives where they are in the sun for very limited time. This can prevent normal social activities and the intense pain that is experience interferes with normal daily activities and can prevent adequate sleep.
Approximately 10,000 people globally are affected by EPP, an estimated 4,000 in the US.
In January 2009 CLINUVEL announced interim results from its lead Phase III study of SCENESSE (afamelanotide 16mg) in patients diagnosed with EPP (CUV017). The data from the first 14 Swiss patients to complete the 12 month study period were analysed, showing SCENESSE was of clinical benefit in EPP. For more information, see the company's announcement.
In December 2009 CLINUVEL announced preliminary results from its lead Phase III study of SCENESSE in 100 patients diagnosed with EPP (CUV017). For more information, see the company's announcement.
CLINUVEL released full results from the CUV017 study in July 2010. For more information, see the company's announcement.
In November 2011 CLINUVEL announced results from it first US Phase II study of SCENESSE (CUV030). You can read the results here.
In December 2011 CLINUVEL announced results from its second Phase III study of SCENESSE (CUV029). You can view the results here.
In November 2013 CLINUVEL announced results from its US Phase III study of SCENESSE (CUV039). You can read the results here.
Regulatory status
SCENESSE (afamelanotide 16mg) has been granted Orphan Drug Designation by the EMA, FDA, TGA and Swissmedic for EPP.
In May 2010, the Italian Medicines Agency allowed for the prescription and reimbursement of SCENESSE (afamelanotide 16mg) under Law 648/96 for Italian patients diagnosed with EPP. For more information, see this page.
In December 2014, the European Commission approved SCENESSE to prevent phototoxicity in adult patients diagnosed with EPP, following a recommendation from the European Medicines Agency's Committee for Medicinal Products for Human Use (CHMP). You can read the announcement here.
CLINUVEL is currently working to make SCENESSE available across Europe. If you have EPP and would like to receive updates on our program, please contact us.
References
·Harms JH, et al. Mitigating photosensitivity of erythropoietic protoporphyria patients by an agonistic analog of alpha-melanocyte stimulating hormone. Photochem Photobiol. 2009 Nov-Dec;85(6):1434-9.
·Murphy GM. Diagnosis and Management of the Erythropoietic Porphyrias, Dermatologic Therapy 2003;16:57-64.
·Thunell S, Harper P, Brun A. Porphyrins, Porphyrin Metabolism and Porphyrias. IV. Pathophysiology of Erythropoietic Protoporphyria - Diagnosis, Care and Monitoring of the Patient. Scand J Clin Lab Invest 2000;60:581-604.
·Todd DJ. Clinical Implications of the Molecular Biology of Erythropoietic Protoporphyria, J Eur Acad Dermatol Venerol 1998;11:207-13.
New Positive Clinical Results for Givosiran (ALN-AS1) in Acute Intermittent Porphyria Patients with Recurrent Attacks
Wednesday - August 9, 2017 @ 07:00:23
26 Jun, 2017New Positive Clinical Results for Givosiran (ALN-AS1) in Acute Intermittent Porphyria Patients with Recurrent Attacks
We reported interim results from our ongoing Phase 1 study with givosiran at the 2017 International Congress on Porphyrins and Porphyrias (ICPP), held June 25 28, 2017 in Bordeaux, France. Data presented were from the first three unblinded cohorts from Part C, in patients with acute intermittent porphyria (AIP) that experience recurrent attacks and initial data from the open-label extension (OLE) study.
Patients treated with givosiran (N=9) experienced a mean 63 percent reduction in the annualized number of all porphyria attacks relative to the run-in period attack rate, with consistent effects observed across a wide range of baseline attack rates. Evaluating only attacks that were treated at a healthcare facility or with hemin, givosiran administration was associated with a mean 73 percent reduction in annualized attack rate relative to placebo during the treatment period. A 73 percent mean decrease in annualized hemin doses relative to the run-in period was also reported. Additionally, in a new analysis, the observed reduction in annualized attack rate was found to be associated with the degree of ALA and PBG lowering.
Further, initial results from Cohorts 1 and 2 (N=8) of the givosiran OLE study were also presented; to date, all eligible patients have rolled over from the Phase 1 study to the OLE study. These data showed that that longer-term treatment with givosiran was associated with consistent reductions in the annualized porphyria attack rate.
Importantly, as of the data cutoff date, givosiran administration was generally well tolerated in recurrent attack AIP patients in Cohorts 1-3 in Part C of the Phase 1 study and in Cohorts 1 and 2 of the ongoing OLE study, with a mean of 169 and 111 days on study, respectively, and up to 12 months on givosiran. In Part C there were no drug-related serious adverse events (SAEs) or discontinuations due to adverse events (AEs). Excluding porphyria attacks, three patients had four SAEs; none were assessed as related to study drug. As previously reported, one death occurred in a patient in cohort 3 in the givosiran arm due to hemorrhagic pancreatitis complicated by a pulmonary embolism and following a recent hospitalization for bacteremia; the death was considered to be unlikely related to study drug by the investigator and the studys Safety Review Committee. During the Phase 1 treatment period, all randomized patients reported at least one AE.
The majority of AEs were assessed as mild or moderate in severity. Twenty-five percent of patients had severe AEs, assessed as unrelated to study drug. AEs in three or more patients included: abdominal pain, headache, nasopharyngitis, nausea and vomiting. Four patients were assessed as having AEs possibly related to study drug, including injection site reaction (mild and self-limiting), hypersensitivity, myalgia, headache, moderate renal impairment (in a patient with a history of moderate renal impairment) and erythema. There were no other clinically significant changes in vital signs, electrocardiograms, clinical laboratory parameters (including liver function tests and lipase tests), or physical examination. The overall safety experience in the ongoing OLE study was consistent with results from the Phase 1 study. No SAEs (excluding porphyria attacks) or discontinuations due to AEs have been reported in the OLE study.
Separately, updated 12-month data from EXPLORE a prospective, multinational, observational study characterizing the natural history and clinical management of acute hepatic porphyria (AHP) patients with recurrent attacks or who receive hemin prophylaxis to prevent attacks demonstrate that patients suffer from both acute attacks and chronic symptoms in between attacks, that together result in a diminished quality of life. The annualized attack rate on study was approximately 5 attacks per person with a mean attack duration of 7 days. The majority of attacks (77 percent) required treatment in the hospital, urgent healthcare facility or with intravenous hemin.
Further, in the first analysis of its kind for AHP in the U.S., an analysis of direct costs associated with AHP and recurrent attacks revealed the average estimated annual expenditure per patient ranges from approximately $400,000 to $650,000 of direct costs.
We believe these latest datasets further support givosirans potential to transform the treatment of patients suffering from acute hepatic porphyrias, and we look forward to continuing our efforts in rapidly developing givosiran towards regulatory filings and, if approved, to patients.
Holly Hamilton joins the APF CEP Facebook group
Friday - August 4, 2017 @ 15:57:40
Welcome Holly Hamilton as our new Moderator the the Closed Facebook CEP group. Holly and her husband Justin are wonderful parents and folks. Justin & Holly are the proud parents of 2 adorable children.
They also support the American Porphyria Foundation and spreading awareness. To learn more about Justin & Holly and how they manage Justins CEP together catch the interview at this link: http://www.porphyriafoundation.com/content/justin-hamilton To learn more about CEP: porphyriafoundation.org To join the CEP FB Group: https://www.facebook.com/groups/Apf.CEP/
Thank you for supporting the APF!
IMPORTANT NEW RESEARCH STUDY FOR PCT
Friday - August 4, 2017 @ 07:00:06
IMPORTANT!!! A new research study is starting for PCT. The Harvoni Treatment for Porphyria Cutanea Tarda study is Beginning to recruit patients now. Please contact the APF office on 1-866-APF-3635 to find out how to participate.
Remember~ "Research is your key to a cure"
APF Membership
Tuesday - August 1, 2017 @ 06:00:18
APF Membership
APF Membership: Guess what? It's FREE!
DNA is FREE to all who join the Longitudinal Study!
Doctor packets are FREE!
Patient packets are FREE!
Subscription to our Weekly E-news is FREE!
Consults by experts with your doctor is FREE!
Brochures are FREE!
Hope is FREE!
And so much more!!!
Join the APF.it's FREE!
Is your membership and contact information up to date? If you have moved, please update us with your current address. Also, be sure to send us your email address so you can receive the weekly Porphyria Post.
Donations and/or special contributions are appreciated:
A Memorial or Remembrance Gift honors a loved one or friend as it provides support for vital porphyria research and educational programs.
Protect the Future Fund: The lack of future porphyria specialist is a crisis. There is no special government funding to train new porphyria specialist, so we must do this ourselves. Without financial support, we run the risk of losing knowledge of the disease, quality testing, diagnosis and treatment.
Research projects
Patient and Physician education
To make a contribution, please use the links provided or call the APF at 1.866.APF.3635 or 713.266.9617!
Claire Sadowniczak of Orlando, Florida, is a member of the APF. She keeps the group encouraged and laughing. Her stories about her turtle Alamo are sheer delight and lessons in tenacity for all of us.
My mother and I have AIP; I started attacks at age 11. One thing that gives me great pleasure is rescuing a wild animal, nursing it back to health and releasing it back into the wild. One rescued 5" brown and black Florida mud turtle left me a present, an egg. It hatched on a freezing day, so I couldn't release it. The black hatchling was smaller than a dime, the shell still soft when I picked her up. I carried her in the palm of my hand as I was preparing a tank of gravel with a plastic sour cream lid as her "pond". We went to the pet store to try to find food small enough for her. They said I'd never keep her alive and she's now 11 years old. When I finally put her into her tank, she ran to the front glass begging me to pick her up again. She ignored my screaming Pomeranian who was jumping up and down in front of her tank. I named her Alamo for her courage in the face of danger.
Water turtles do not have salivary glands or pulmonary muscles to swallow air to breathe. After being out of the water awhile, her mouth dries out and her beak "squeaks". When this occurs, I put her back into her pond. Once I picked her out of her pond and put her on hubby's lap, so I could clean her tank. She doesn't like him so she began squeaking her beak when I left the room, begging to get off his lap!
She potty-trained herself, too. I kept a towel on my lap when I held her. She realized that I didn't like her mess and hasn't had an accident since she was six months old. She just squeaks her beak to ask to go back into her tank.
I feed her with a plastic spoon. Since she is in the snapping turtle family, I didn't want her to associate fingers with food. She'll swallow anything presented on a spoon, including medicine and will follow a spoon anywhere! She also loves TV and gets very involved. If a car explodes on a show, she'll open her mouth at the TV.
Once she had a respiratory infection; turtles can catch colds from people. The vet told me to add a heater and thermometer to her tank to keep the water at 80 degrees. She did not like either, popping the heater off its rubber suction cups and bashing it and the thermometer against the rocks till I removed them. It is HER home after all.
Alamo enjoys being an only child and will attack a mirror until it is removed. Although the breed is supposed to be "vicious", she is a sweety with me. She once saw a piece of shiny fuzz on my sweater that she wanted to eat. She very gently tried to get it with the side of her mouth so she would not accidentally bite me through the sweater.
I carry her around under the hem of my T-shirt or in a pocket for hours, and her little head comes out like a periscope to look around. She sits on my lap in the car. In the vet's office, Alamo watches the other animals from my lap and feels quite safe and content. I put her on the grass in the back yard for exercise, and she runs straight back to me. Definitely a lap turtle!
Alamo's Mother, Claire Sadowniczak Prior to my diagnosis of Acute Intermittent Porphyria, I underwent abdominal surgery with sodium pentothal, went into cardiac arrest and a three month major porphyria attack. Therefore, when I learned that I needed gallbladder surgery, I was very nervous.
I found a general surgeon, Dr. Cesar Cabascango, who is not only familiar with porphyria, but I was his fifth patient with porphyria. Normally, with the laproscopic gallbladder procedure, you have surgery the same day as admission and go home the following morning. Because of my porphyria, Dr. Cabascango had me admitted the day before surgery, opened a central line with three ports in my chest, and infused large doses of dextrose. He continued to infuse dextrose during surgery and for an additional two and a half days in the hospital after surgery. By faxing information on porphyria to my insurance company, he even got them to approve the additional stay in the hospital. I did not suffer a porphyria attack whatsoever.
The surgery was performed at Florida Hospital Orlando. The Assistant Director of Anesthesiology handled my case personally, and brought me through the procedure with no problems. Everyone at the hospital researched porphyria, read the brochures from the American Porphyria Foundation that I provided, questioned me about it, and treated me with such special care that it was my most positive hospital experience ever.
Many people with porphyria have horror stories about past medical care, including myself, but things are improving and the brochures provided by the American Porphyria Foundation are a great benefit when distributed to health care professionals.
Keep Yourself Moving!
Monday - July 24, 2017 @ 06:00:26
Keep Yourself Moving
If exercise were a pill, it would be the most widely prescribed medication in the world. (Emory University School of Medicine) Of all the things we can do for our health, few are more generally helpful than physical exercise.
Exert yourself. Leading a physically active life can help us feel happier, think more clearly, have more energy, be more productive and, along with proper diet, control our weight. Exercise need not be painful or extreme to be effective. Regular periods of moderate exercise several times a week can be very beneficial.
Jogging, brisk walking, biking, and taking part in active sportsenough both to get your heart beating faster and to cause you to break a sweatcan improve your endurance and help to prevent heart attack and stroke. Combining such aerobic exercise with moderate weight training and calisthenics helps to strengthen your bones, internal muscles, and limbs. These activities also contribute to maintaining a higher metabolism, which automatically helps to control your weight.
Exercise can be enjoyable
Use your feet. Exercise is beneficial for people of all ages, and membership in a gym is not required to get it. Simply using your feet instead of a car, bus, or elevator is a good start. Why wait for a ride when you can walk to your destination, perhaps even arriving there faster? Parents, encourage your children to participate in physical play, outdoors whenever possible. Such activity strengthens their bodies and helps them to develop whole-body coordination in ways that sedentary entertainment, such as video games, cannot.
No matter how old you are when you start, you can benefit from moderate physical exercise. If you are older or have health problems and have not been exercising, it is wise to consult a doctor about how to begin. But do begin! Exercise that is started gradually and not overdone can help even the oldest among us to maintain muscle strength and bone mass. It can also help seniors to avoid falls.
Exercise is what helped Rustam, mentioned in the first article of this series. Seven years ago, he and his wife began jogging a little each morning, five days a week. At first, we made excuses not to go, he relates. But having a partner helped to motivate both of us. Now it has become a good, enjoyable habit.
Rocco shares his story with EPP
Friday - July 21, 2017 @ 06:00:27
Sometimes the pain takes hold of the psyche and then takes over your life completely. At such moments, I have often wished that my illness could somehow be more apparent to others. Sometimes when I am treated with condescension because the pain which feels like boiling oil on my skin is not visible, I think about people who are blind or paralyzed. Nobody doubts a persons disability when they see them with a white stick or a wheelchair.
Rocco Falchetto can now enjoy being outside in the sunshine without worrying about his skin burning after a few moments exposure. (Photo provided by the author)
Thoughts like this are irrational of course I am thankful that I dont suffer from the additional problem of a visible disability. Despite this, the disease has a significant effect on my quality of life and it is hard to get away from the negative mindset especially when doctors downplay the symptoms because they don't know what they are talking about or as a result of their lack of specialist knowledge about the illness. I always get angry when I hear similar stories from other sufferers.
While I would like to experience more understanding and sympathy for my condition, I can't stand pity at all. I have the disease, but Rocco Falchetto is not the disease he is much more than that. While my illness has partly shaped who I am today, I still lead a fulfilling life and have achieved things that I can be proud of. This is another reason why it is so difficult for me to talk about it. People should get to know me first without knowing anything about the illness.
However, I now know just how important it is to tell my story.
It is high time that society takes the needs of sufferers of rare illnesses and diseases into account and the difficulties we face in finding treatments.
A difficult childhood
EPP meant that my childhood and teenage years were difficult. The many associated problems and limitations were difficult to accept. The desire to be normal was often much greater than common sense dictated, and put me in situations that led to a lot of pain and suffering.
The situation was also difficult for my parents, especially as one of my two older brothers also suffered from EPP. They tried their hardest to support us in leading as normal a life as we could. Of course, this was not always possible. I still remember their helplessness when my brother died of liver complications at the age of 16 in 1974, something which can occur to EPP patients in rare cases. Back then, the doctors could do nothing for him. Nowadays, his life could have been saved by a liver transplant and immunosuppressants.
The thought that this might happen to me as well together with the need for such a complicated operation is always at the back of my mind and becomes apparent in certain situations. For example, I am always nervous as to whether all my liver values are normal after every check-up.
Sometimes they deviate from the norm, meaning additional tests are then necessary to make sure that they dont deteriorate further. In such moments, I often think about my brothers death and what my parents had to go through at the time the hospital visits, the trip by ambulance to the University Childrens Hospital in Zurich and the moment that the doctors announced they could no longer help him. I was too young to really understand what was happening back then. However, I clearly remember my mothers devastation at losing her child and the feeling of helplessness and injustice.
The long path to acceptance
After living with EPP for many years and unsuccessfully trying out various remedies and products in an attempt to alleviate the debilitating symptoms, I finally began to accept the illness.
Whenever possible, I didnt leave the house at all on sunny days. When going to work, for example, I was always afraid of the painful symptoms of EPP. I walked in the shade as far as possible, wore long-sleeved shirts and gloves, and even covered my face in extreme cases.
However, keeping up these precautionary measures for long periods was not realistic as it would have led to almost complete social isolation. I then had to expose myself to dangerous situations from time to time, even though I knew about the possible adverse effects that may have occurred as a result.
Hopes for a normal life
The turning point for me came when I first learned about research by another company into a substance produced naturally in the body that stimulates tanning of the skin. I discovered it online completely by accident at a time when it was still being tested in other applications. I saw its potential in treating EPP and talked to my doctor. Within a matter of months, she and the researchers concerned had organized the first human clinical trial for it.
I was one of five patients in Switzerland who took part in the clinical study.
I will never forget the day when I first received the drug I summoned all my courage and went to sit in the sun. I then waited, afraid of what was going to happen. Ten minutes passed, then 20, then 30 (where I would normally already be in pain), then 40 minutes and more in the sun and all without the typical painful symptoms! After over 40 years with the illness, I was convinced that I had finally found a drug that I believed tackled the EPP symptoms.
I took part in a further clinical trial. I had to travel to Zurich every two months for a new injection of the drug implant, which was a small price to pay given what I felt was its positive effect on my quality of life.
I am currently benefitting from a compassionate use program which allows me to continue using the drug and, thanks to special legislation, most Swiss health insurance companies now reimburse the cost of such medication.
Difficult regulatory environment
However, the current situation is only temporary if the product is not formally approved, then the reimbursement and the treatment can be withdrawn. This would be a heavy blow.
I dont like to think about what would happen in this case, but the increasingly challenging context surrounding drug approvals does make me nervous as a patient.
Some countries have even started to put a price on the quality of life. For example, at the end of 2010 the Swiss Federal Court passed a judgment ruling that health insurance companies were not obliged to reimburse the costs for treating Pompe disease, a rare, serious metabolic disorder. Treatment of Pompe disease can cost several hundred thousand Swiss francs a year. While this is undoubtedly expensive, the therapy can significantly ease the symptoms and allow patients to lead a relatively normal life.
This shows how difficult it still is for people with rare diseases to make their voices heard, not to mention how inadequately their needs continue to be met. I am also worried that our modern society is not more committed to discovering creative and innovative solutions for the benefit of these patients.
This medication has radically changed my day-to-day life I can now do things that I would never have been able to do before without pain. Not only my friends and family benefit from this my working life has also become much easier.
I can now go to the office without having to always move about in the shade and go on business trips without thinking so much about the unfamiliar surroundings. Lastly, I can now move freely around at work, meet with colleagues and simply enjoy our wonderful working environment.
The EPP sufferer community is a small one in Switzerland, gathered in a patient organization that I chair. We all know one another. It is very moving to hear how patients who have undergone treatment with the drug felt the sun warm their faces for the very first time without causing pain and suffering.
Organizations around the world unite for Porphyria links
Wednesday - July 19, 2017 @ 06:00:00
The following organizations have websites in English
The American Porphyria Foundation has a very extensive website, including a section for medical staff: www.porphyriafoundation.com
Existen dos tipos principales de porfirias: Uno es el que afecta la piel (cutáneo) y el otro es el que afecta el sistema nervioso. Las personas que tienen porfiria cutánea desarrollan ampollas, picazón e inflamación en la piel cuando se exponen al sol. El tipo de porfiria que afecta al sistema nervioso se llama porfiria aguda. Los sÃntomas incluyen dolor en el pecho, abdomen, brazos o piernas, espalda, adormecimiento de los músculos, hormigueo, parálisis o calambres, vómitos, estreñimiento y cambios mentales o en la personalidad. Estos sÃntomas pueden aparecer y desaparecer.
NIH: Instituto Nacional de la Diabetes y las Enfermedades Digestivas y Renales
https://medlineplus.gov/spanish/porphyria.html
What are common signs and symptoms of AHP?
Wednesday - July 12, 2017 @ 14:44:31
What are common signs and symptoms of AIP?
Severe abdominal pain: the most common AIP symptom
The most common symptom of AIP is severe abdominal pain that usually cannot be relieved with pain medicine such as Advil (ibuprofen) or Tylenol (acetaminophen). More than 85% of people who develop AIP symptoms have abdominal pain.
Experiencing symptoms is known as having an AIP attack. Symptoms may occur for a set period of time, then go away only to come back later.
Common AIP symptoms
Symptoms can occur in many different areas of your body during an AIP attack. These include:
GASTROINTESTINAL (GI) SYMPTOMS
Abdominal pain
Vomiting
Constipation
Diarrhea
GASTROINTESTINAL (GI)
URINARY SYSTEM
BRAIN OR NERVOUS SYSTEM
HEART OR BLOOD VESSELS
Early diagnosis and treatment of AIP are critical
AIP attacks can be very serious. And symptoms may get worse over time. Untreated attacks can cause serious damage to your nervous system including paralysis, and even death. That's why early diagnosis and treatment of AIP is so important.
If you have any of the symptoms listed above, talk to your doctor right away.
What triggers AIP attacks?
Steroid hormones, particularly estrogen and progesterone. These hormones fluctuate the most during the 2 weeks before a womans menstrual periods start.
Unhealthy behaviors such as drinking alcohol, smoking, or using illegal drugs.
Stress on the body caused by infections, surgery, or physical exhaustion.
Certain prescription drugs. Attacks can also be triggered by starting a new prescription drug.
Changes in eating patterns such as fasting or crash dieting.
AIP is caused by a partial lack of an enzyme known as porphobilinogen deaminase (PBGD). An enzyme is a type of protein that helps to regulate the bodys tissues and organs. Enzymes carry out almost all of the thousands of chemical reactions that take place in cells.
If you have AIP, you have about half of the normal amount of PBGD in your body. This is usually enough for your body to do what it is supposed to do. But triggers like those listed above can upset your body's chemical balance enough to cause symptoms.
Important Safety Information PANHEMATIN should only be used by doctors experienced in the management of porphyrias in hospitals where the recommended clinical and laboratory diagnostic and monitoring practices are available.
TAKE STEPS TO HELP MANAGE YOUR AIP:
Try to identify your possible triggers. Then try to reduce or avoid as many as you can. If you continue to experience attacks, keep writing down suspected triggers. Look for patterns of things that occurred right before an attack to identify any changes you can make. Talk to your doctor if you need help.
Be careful when changing your eating patterns. If you want to lose weight, get advice from your doctor or nutritionist before starting any diet.
Call your doctor at the first sign of an attack. Repeat attacks are often similar. They may start the same way and you may have the same symptoms.
Wear a medical alert bracelet. Doctors need to know about your AIP so that they do not prescribe drugs that may make your AIP worse.
Indications And Usage
PANHEMATIN is a hemin for injection prescription medication used to relieve repeated attacks of acute intermittent porphyria related to the menstrual cycle in affected women, after initial carbohydrate therapy is known or suspected to be inadequate.
Limitations of Use
Before giving PANHEMATIN, consider an appropriate trial of carbohydrates [i.e., 400 grams of glucose (a carbohydrate or sugar) per day for 1 to 2 days].
Attacks of porphyria may progress to a point where irreversible nerve damage has occurred. PANHEMATIN therapy is intended to prevent an attack from reaching the critical stage of nerve breakdown. PANHEMATIN is not effective in repairing nerve damage that has already occurred.
Important Safety Information
PANHEMATIN is not for patients known to be allergic to this drug.
Vein inflammation is possible. Use a large arm vein or a central line to administer PANHEMATIN to minimize the possibility of vein inflammation.
Elevated iron levels may occur. Your doctor must monitor your iron levels if you receive multiple courses of PANHEMATIN.
PANHEMATIN has a passing and mild blood thinning effect. Avoid blood thinners during PANHEMATIN therapy.
Reversible kidney shutdown has occurred when too high a dose of PANHEMATIN was given in a single dose.
PANHEMATIN may carry a risk of transmitting agents that can cause infections, such as viruses, and theoretically, the Creutzfeldt-Jakob disease (CJD) agent.
Most common side effects are headache, fever, infusion site reactions, and vein inflammation.
To report SUSPECTED SIDE EFFECTS, contact Recordati Rare Diseases Inc. at 1-888-575-8344, or FDA at 1800-FDA-1088 or www.fda.gov/medwatch.
When taking PANHEMATIN, do not take drugs such as estrogens (e.g., oral contraceptives), barbiturates (drugs that help with sleep and used to treat epilepsy) or steroids (body hormone-like drugs), because such drugs can trigger an attack or make an attack worse.
Although these features may also occur on the face and neck as well, it is more common to notice mottled brown patches around the eyes and increased facial hair (hypertrichosis). Occasionally the skin becomes hardened (scleroderma) on the neck, face or chest. There may be small areas of permanent baldness (alopecia) or ulcers.
Characteristically, the urine is darker than usual, with a reddish or tea-coloured hue.
The clinical appearance of variegate porphyria is similar but the biochemical abnormality is different.
Porphyria cutanea tarda
What is the cause of porphyria cutanea tarda?
PCT is due to a defective enzyme in the liver (uroporphyrinogen decarboxylase). This is involved in synthesis of the red pigment in blood cells (haem). Some families carry an enzyme that is prone to oxidation under certain circumstances, most often due to iron accumulation.
There are basically two forms of PCT, type 1 and type 2.
Type 1 PCT generally begins in mid-adult life after exposure to certain chemicals that increase the production of porphyrins (precursors of haem) in the liver. These include:
oestrogen eg oral contraceptive, hormone replacement (in 50% of affected women) or liver disease
polychlorinated aromatic hydrocarbons (eg, dioxins, when PCT is associated with chloracne).
iron overload, due to excessive intake (orally or by blood transfusion), viral infections (hepatitis, especially hepatitis C, in 15%) or chronic blood disorders such as thalassaemia (acquired haemochromatosis), or hereditary haemochromatosis (in 20%)
Type 2 PCT is familial and associated with abnormal genetic variants of uroporphyrinogen decarboxylase. Trigger factors are less often involved and onset of PCT is often younger than in type 1 PCT.
What investigations should be done?
Skin biopsy: characteristic changes are seen which differentiates PCT from other blistering diseases.
Examination of the urine with a Wood's lamp: may reveal coral pink fluorescence due to excessive porphyrins.
24 hour urine porphyrin profile: total porphryins are usually elevated 5- to 20-fold above the upper reference limit. Most of the excreted porphyrins are uroporphyrin and a 7-carboxyl porphyrin.
Other important tests include:
Complete blood count to assess haemoglobin levels.
Measurement of iron stores, which may be increased in over 30% of patients.
Liver enzymes because the liver sometimes does not function normally.
Fasting blood sugar because of the increased incidence of diabetes.
Antinuclear antibodies because of the increased incidence of lupus erythematosus.
Viral hepatitis studies
If a blood ferritin shows iron stores are increased, further investigations may include transferrin saturation and genotyping for hereditary haemochromatosis.
What is the management of porphyria cutanea tarda
Avoid alcohol, oestrogen, and iron.
If using pesticides, be very careful to avoid contact with polychlorinated aromatic hydrocarbons (eg. 2,4,5T).
Apply an opaque sun-block and cover up when outside; the responsible light is the "Soret" band at 400 nm which is unfortunately not blocked by most sunscreens.
Use tanning cream containing dihydroxyacetone: this can block the responsible light to some extent.
Phlebotomy (removal of blood) up to 500 ml blood is removed every one to two weeks until the haemoglobin and iron levels drop to low normal levels. It may take 3 to 6 months to improve. Venesection may need to be repeated after a year or more.
Antimalarial tablets, ie, low-dose chloroquine or hydroxychloroquine may be recommended, but must be used cautiously. This medication makes the porphyrins more soluble so more are excreted in the urine.
Autologous red cell transfusion (this is a blood transfusion using the patient's own red cells that have been washed to remove plasma thereby reducing the circulating porphyrins).
In late 2008, after a yearlong battle, I was diagnosed with acute intermittent porphyria (AIP). At first, I was relieved to finally have answers. Then, I learned how little there was that could be done with the disorder. I wasn't the first in my family to be diagnosed. In the late 1950's my great uncle was diagnosed and then died of the disorder. My grandmother would follow in the early 60's at the young age of 28. My mother was also diagnosed in the mid 1990's. Needless to say I thought I understood the seriousness of the situation, but after learning what my treatment options were, I became nervous. After all, I'm married and have two children. How was my ability to support them going to be affected? I did some research on the disorder and stumbled across the American Porphyria Foundation. Through conversing with them I learned of some opportunities to help find better treatments and, maybe, a cure. I jumped at the chance with more concern about my children having the gene than with concern about myself.
The first study I enrolled in was simple and straight forward. It required no travel and only one time did I have to send them any samples. It was simply blood which was drawn at my local doctors office and then mailed off to Salt Lake City. The Longitudinal Study is a simple study and I strongly encourage everyone to take part in this study. It was through this study that I was able to get gene testing done at no cost to me. Now, I have concrete proof of my diagnosis. I am still in the study and once in a great while fill out more papers with questions about how I am doing. Like I said-- simple and straight forward.
The second study I took part in was the Panhematin study. This study required me to fly to Galveston, Texas and stay there in the hospital for four days. The research grant paid for all of my travel expenses and there was of course no in hospital expenses for me to worry about. The APF arranged all the travel. Again, this study had a simple procedure. Once I began feeling as though I had an attack coming on I contacted the foundation and they made the travel arrangements. The next day I flew into Houston Hobby Airport where a car service picked me up and drove me to University of Texas Medical Branch in Galveston. This particular study was a double blind study. That means that neither I, nor the doctors and nurses know if I was getting the Panhematin or a placebo. Only the pharmacist knew. In order to keep the secrecy I had to wear a blindfold, the tubing and medicine was wrapped in foil, and a sheet was also placed between me and the bottle of whatever I was getting. This happened once a day for four days, and I was also on normal saline with 10% dextrose constantly while I was there. In all my experience with this study was good. Things went smoothly and before I knew it, I was on my way back home to Colorado.
Data Presented at ICPP Show Encouraging Results for Givosiran in AHP Patients
Tuesday - June 27, 2017 @ 12:09:59
Data Presented at ICPP Show Encouraging Results for Givosiran in AHP Patients
Mathew Shanley
Published Online: Monday, Jun 26, 2017
200Reddit0
At the 2017 International Congress on Porphyrins and Porphyrias (ICPP) this week in Bordeaux, France, Alnylam Pharmaceuticals is presenting new data on Givosiran (ALN-AS1) for the treatment of acute hepatic porphyrias (AHP).
In a randomized, double-blind, placebo-controlled study, Givosiran demonstrates a decreased annualized attack rate and hemin usage. Initial results from an ongoing open-label extension (OLE) study show consistent reductions in porphyria attacks with continued Givosiran treatment.
Hepatic porphyrias are a subgroup of of porphyrias in which the enzyme deficiency occurs in the liver. Acute hepatic porphyrias encompass four diseases: acute intermittent porphyria (AIP) (most common), variagate porphyria, hereditary coproporphyria, and hereditary deficit of delta-aminolevulinic acid dehydratase (most rare).
Currently, urgent treatment with an injection of human hemin and/or perfusion of carbohydrates is required when an acute attack is confirmed. Management of the disorder includes avoiding causal factors to prevent attacks and protect the skin from the light in cases of cutaneous manifestations.
Alnylams hope in developing Givosiran, a subcutaneously administered, investigational RNAi therapeutic, is for it to become a novel treatment approach for the prevention of recurrent attacks by targeting ALAS1 for the treatment of AHP, including AIP.
"We believe that a long acting therapeutic agent that has the potential to prevent porphyria attacks and that can be administered via a once monthly, low volume, subcutaneous injection could be a potentially transformative treatment option for patients suffering with this debilitating and potentially life-threatening disease," said Jeff Miller, General Manager of the givosiran program.
"Based on these encouraging interim results and with both Breakthrough Therapy and PRIME designations granted, we will continue to work with global regulatory authorities to rapidly advance givosiran toward regulatory filings and, if approved, to patients. To that end, we remain on track to initiate the givosiran Phase 3 program in late 2017."
Per a press release, the continuing part of the Phase 1 study of the drug is being managed as a randomized, double-blind, placebo-controlled study in up to 24 AIP patients who experience recurrent porphyria attacks. Patients are followed in a 3-month run-in phase, where the amount and frequency of attacks and levels of porphobilinogen (PBG) and aminolevulinate (ALA) are measured prospectively.
Patients who experience at least 1 porphyria attack during the run-in phase are then eligible to enter the studys 6-month treatment phase, where they are randomized to receive 2 once-quarterly doses or 4 once-monthly doses of placebo or givosiran at 2.5 or 5.0 mg/kg. During the treatment phase, the effects of placebo or givosiran on the number and frequency of porphyria attacks, as well as on the levels of ALA and PBG, are measured prospectively in a blinded manner and then compared to run-in phase results.
Additional measures include safety, tolerability, hospitalizations, use of hemin (FDA approved), levels of ALAS1 mRNA, and givosiran pharmacokinetics. Following the treatment phase, all patients are eligible to receive givosiran in an open-label extension study.
Givosiran has already been granted Orphan Drug Designations in both the E.U. and the U.S. for the treatment of acute hepatic porphyrias. Givosiran also received Breakthrough Designation by the U.S. Food and Drug Administration (FDA), and has been granted PRIME designation by the European Medicines Agency (EMA).
For more updates on orphan drugs, follow Rare Disease Report on Facebook and Twitter.
Here is one to print and share with your Doctors Overview from: Herbert L. Bonkovsky, M.D., Carolinas Health Care System
Monday - June 26, 2017 @ 07:00:31
Porphyria
What are porphyrias?
Porphyrias are rare disorders that affect mainly the skin or nervous system and may cause abdominal pain. These disorders are usually inherited, meaning they are caused by abnormalities in genes passed from parents to children. When a person has a porphyria, cells fail to change body chemicals called porphyrins and porphyrin precursors into heme, the substance that gives blood its red color. The body makes heme mainly in the bone marrow and liver. Bone marrow is the soft, spongelike tissue inside the bones; it makes stem cells that develop into one of the three types of blood cellsred blood cells, white blood cells, and platelets.
The process of making heme is called the heme biosynthetic pathway. One of eight enzymes controls each step of the process. The body has a problem making heme if any one of the enzymes is at a low level, also called a deficiency. Porphyrins and porphyrin precursors of heme then build up in the body and cause illness.
What is heme and what does it do?
Heme is a red pigment composed of iron linked to a chemical called protoporphyrin. Heme has important functions in the body. The largest amounts of heme are in the form of hemoglobin, found in red blood cells and bone marrow. Hemoglobin carries oxygen from the lungs to all parts of the body. In the liver, heme is a component of proteins that break down hormones, medications, and other chemicals and keep liver cells functioning normally. Heme is an important part of nearly every cell in the body.
What are the types of porphyria?
Each of the eight types of porphyria corresponds to low levels of a specific enzyme in the heme biosynthetic pathway. Experts often classify porphyrias as acute or cutaneous based on the symptoms a person experiences:
Acute porphyrias affect the nervous system. They occur rapidly and last only a short time.
Cutaneous porphyrias affect the skin.
Two types of acute porphyrias, hereditary coproporphyria and variegate porphyria, can also have cutaneous symptoms.
Experts also classify porphyrias as erythropoietic or hepatic:
In erythropoietic porphyrias, the body overproduces porphyrins, mainly in the bone marrow.
In hepatic porphyrias, the body overproduces porphyrins and porphyrin precursors, mainly in the liver.
Table 1 lists each type of porphyria, the deficient enzyme responsible for the disorder, and the main location of porphyrin buildup.
The exact rates of porphyria are unknown and vary around the world. For example, porphyria cutanea tarda is most common in the United States, and variegate porphyria is most common in South America.1
What causes porphyria?
Most porphyrias are inherited disorders. Scientists have identified genes for all eight enzymes in the heme biosynthetic pathway. Most porphyrias result from inheriting an abnormal gene, also called a gene mutation, from one parent. Some porphyrias, such as congenital erythropoietic porphyria, hepatoerythropoietic porphyria, and erythropoietic protoporphyria, occur when a person inherits two abnormal genes, one from each parent. The likeliness of a person passing the abnormal gene or genes to the next generation depends on the type of porphyria.
Porphyria cutanea tarda is usually an acquired disorder, meaning factors other than genes cause the enzyme deficiency. This type of porphyria can be triggered by
too much iron
use of alcohol or estrogen
smoking
chronic hepatitis Ca long-lasting liver disease that causes inflammation, or swelling, of the liver
HIVthe virus that causes AIDS
abnormal genes associated with hemochromatosisthe most common form of iron overload disease, which causes the body to absorb too much iron
For all types of porphyria, symptoms can be triggered by
use of alcohol
smoking
use of certain medications or hormones
exposure to sunlight
stress
dieting and fasting
What are the symptoms of porphyria?
Some people with porphyria-causing gene mutations have latent porphyria, meaning they have no symptoms of the disorder. Symptoms of cutaneous porphyrias include
oversensitivity to sunlight
blisters on exposed areas of the skin
itching and swelling on exposed areas of the skin
Symptoms of acute porphyrias include
pain in the abdomenthe area between the chest and hips
pain in the chest, limbs, or back
nausea and vomiting
constipationa condition in which an adult has fewer than three bowel movements a week or a child has fewer than two bowel movements a week, depending on the person
urinary retentionthe inability to empty the bladder completely
confusion
hallucinations
seizures and muscle weakness
Symptoms of acute porphyrias can develop over hours or days and last for days or weeks. These symptoms can come and go over time, while symptoms of cutaneous porphyrias tend to be more continuous. Porphyria symptoms can vary widely in severity.
How is porphyria diagnosed?
A health care provider diagnoses porphyria with blood, urine, and stool tests. These tests take place at a health care providers office or a commercial facility. A blood test involves drawing blood and sending the sample to a lab for analysis. For urine and stool tests, the patient collects a sample of urine or stool in a special container. A health care provider tests the samples in the office or sends them to a lab for analysis. High levels of porphyrins or porphyrin precursors in blood, urine, or stool indicate porphyria. A health care provider may also recommend DNA testing of a blood sample to look for known gene mutations that cause porphyrias.
How is porphyria treated?
Treatment for porphyria depends on the type of porphyria the person has and the severity of the symptoms.
Acute Porphyrias
A health care provider treats acute porphyrias with heme or glucose loading to decrease the livers production of porphyrins and porphyrin precursors. A patient receives heme intravenously once a day for 4 days. Glucose loading involves giving a patient a glucose solution by mouth or intravenously. Heme is usually more effective and is the treatment of choice unless symptoms are mild. In rare instances, if symptoms are severe, a health care provider will recommend liver transplantation to treat acute porphyria. In liver transplantation, a surgeon removes a diseased or an injured liver and replaces it with a healthy, whole liver or a segment of a liver from another person, called a donor. A patient has liver transplantation surgery in a hospital under general anesthesia. Liver transplantation can cure liver failure. More information is provided in the NIDDK health topic, Liver Transplantation.
Cutaneous Porphyrias
The most important step a person can take to treat a cutaneous porphyria is to avoid sunlight as much as possible. Other cutaneous porphyrias are treated as follows:
Porphyria cutanea tarda. A health care provider treats porphyria cutanea tarda by removing factors that tend to activate the disease and by performing repeated therapeutic phlebotomies to reduce iron in the liver. Therapeutic phlebotomy is the removal of about a pint of blood from a vein in the arm. A technician performs the procedure at a blood donation center, such as a hospital, clinic, or bloodmobile. A patient does not require anesthesia. Another treatment approach is low-dose hydroxychloroquine tablets to reduce porphyrins in the liver.
Erythropoietic protoporphyria. People with erythropoietic protoporphyria may be given beta-carotene or cysteine to improve sunlight tolerance, though these medications do not lower porphyrin levels. Experts recommend hepatitis A and B vaccines and avoiding alcohol to prevent protoporphyric liver failure. A health care provider may use liver transplantation or a combination of medications to treat people who develop liver failure. Unfortunately, liver transplantation does not correct the primary defect, which is the continuous overproduction of protoporphyria by bone marrow. Successful bone marrow transplantations may successfully cure erythropoietic protoporphyria. A health care provider only considers bone marrow transplantation if the disease is severe and leading to secondary liver disease.
Congenital erythropoietic porphyria and hepatoerythropoietic porphyria. People with congenital erythropoietic porphyria or hepatoerythropoietic porphyria may need surgery to remove the spleen or blood transfusions to treat anemia. A surgeon removes the spleen in a hospital, and a patient receives general anesthesia. With a blood transfusion, a patient receives blood through an intravenous (IV) line inserted into a vein. A technician performs the procedure at a blood donation center, and a patient does not need anesthesia.
Secondary Porphyrinurias
Conditions called secondary porphyrinurias, such as disorders of the liver and bone marrow, as well as a number of drugs, chemicals, and toxins are often mistaken for porphyria because they lead to mild or moderate increases in porphyrin levels in the urine. Only highnot mild or moderatelevels of porphyrin or porphyrin precursors lead to a diagnosis of porphyria.
Eating, Diet, and Nutrition
People with an acute porphyria should eat a diet with an average-to-high level of carbohydrates. The recommended dietary allowance for carbohydrates is 130 g per day for adults and children 1 year of age or older; pregnant and breastfeeding women need higher intakes.2 People should avoid limiting intake of carbohydrates and calories, even for short periods of time, as this type of dieting or fasting can trigger symptoms. People with an acute porphyria who want to lose weight should talk with their health care providers about diets they can follow to lose weight gradually.
People undergoing therapeutic phlebotomies should drink plenty of milk, water, or juice before and after each procedure.
A health care provider may recommend vitamin and mineral supplements for people with a cutaneous porphyria.
Points to Remember
Porphyrias are rare disorders that affect mainly the skin or nervous system and may cause abdominal pain.
Each of the eight types of porphyria corresponds to low levels of a specific enzyme in the heme biosynthetic pathway.
The exact rates of porphyria are unknown and vary around the world. For example, porphyria cutanea tarda is most common in the United States, and variegate porphyria is most common in South America.
Most porphyrias result from inheriting an abnormal gene, also called a gene mutation, from one parent.
Porphyria cutanea tarda is usually an acquired disorder, meaning factors other than genes cause the enzyme deficiency.
Symptoms of cutaneous porphyrias include
oversensitivity to sunlight
blisters on exposed areas of the skin
itching and swelling on exposed areas of the skin
Symptoms of acute porphyrias include
pain in the abdomen
pain in the chest, limbs, or back
nausea and vomiting
constipation
urinary retention
confusion
hallucinations
seizures and muscle weakness
A health care provider diagnoses porphyria with blood, urine, and stool tests.
Treatment for porphyria depends on the type of porphyria the person has and the severity of the symptoms.
References
Clinical Trials
The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and other components of the National Institutes of Health (NIH) conduct and support research into many diseases and conditions.
What are clinical trials, and are they right for you?
Clinical trials are part of clinical research and at the heart of all medical advances. Clinical trials look at new ways to prevent, detect, or treat disease. Researchers also use clinical trials to look at other aspects of care, such as improving the quality of life for people with chronic illnesses. Find out if clinical trials are right for you .
What clinical trials are open?
Clinical trials that are currently open and are recruiting can be viewed at www.ClinicalTrials.gov .
This content is provided as a service of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), part of the National Institutes of Health. The NIDDK translates and disseminates research findings through its clearinghouses and education programs to increase knowledge and understanding about health and disease among patients, health professionals, and the public. Content produced by the NIDDK is carefully reviewed by NIDDK scientists and other experts.
The NIDDK would like to thank: Herbert L. Bonkovsky, M.D., Carolinas Health Care System
Glass Beaker Publishing Disa McCarthy EPP Book Project Scope ***Attention EPP PATIENTS***
Friday - June 23, 2017 @ 07:00:27
Glass Beaker Publishing
Disa McCarthy EPP Book Project Scope
December 8, 2016
Overview
1.EPP Book Project Background and Description
The author and interviewer has EPP and is creating a book she wished she could find while researching her own symptoms.
The book content will include basic information about EPP (Erythropoetic ProtoPorphyria), but the bulk of it will be anecdotal stories of the lives of several (6-10) people who have been diagnosed with EPP. It will also include charts of raw, anonymous data from 12-50 people such as the age of diagnosis, severity of symptoms, etc.
2.Implementation
The author will conduct interviews with anyone willing to tell their story. The approximate steps will be:
1)Person with EPP expresses interest in being interviewed
2)Person with EPP is given a survey with basic medical information questions and is asked for available dates to speak via phone/skype.
3)Upon completion of the survey and interview, the data from the survey is retained in a database and the interview transcribed.
3.Deliverables
Survey (see below)
Verbal Interview (see questions below)
Book final product
4.People/Partners involved
Disa McCarthy author/interviewee
Glass Beaker Publishing, LLC Publishing company
American Porphyria Foundation Seeking partnership, specifically help finding people with EPP and supporting the book as a resource upon completion.
People with EPP to complete the survey, to interview, and ultimately as a target market.
5.Timeline/Schedule
The survey can be sent out the first week of January, and interviews will be scheduled for January, February and March of next year. The book will be written by June of 2017, and edited/finalized by August 2017.
Approval and Assistance to Proceed
I am seeking approval, authority (if necessary) and assistance from the American Porphyria Foundation, and welcome any questions, concerns or comments you may have.
Survey
This survey will be given to anyone and everyone who has or thinks they may have EPP. The format will be a Google Form for ease of use and collection. Each question will have the answers available in a multiple-choice format.
Questions:
1)Age at symptom onset?
2)Did you have difficulty getting a diagnosis?
3)How many tests did you undergo to achieve diagnosis?
4)How many doctors did you visit for your EPP symptoms prior to diagnosis?
5)Were you able to attain a diagnosis?
6)Age at diagnosis?
7)Age now:
8)Ethnic background:
9)Does anyone else in your family have EPP that you know about?
10)Does anyone else in your family have porphyria that you know about?
11)Symptoms possessed upon sun exposure: (tingling on chin) (pain) (itching) (blisters) (swelling) (fatigue) (panic)
12)If you have swelling, please rate the level of swelling from 1 5, where 1 is mild, 2 is noticeable, and 5 is skin-splitting.
13)If you have blistering, please rate the level of blistering from 1 5, where 1 is a slight rash to 5, 2nd degree burn level.
14)If you have pain upon exposure, please rate the level of pain from 1 10, where 1 is uncomfortable, 6 is unable to concentrate well through the pain and 10 is unbearable.
15)Where does pain occur upon exposure please list 1 for worst, 2 for 2nd worst, 3 for 3rd worst, etc: (hands) (arms) (face) (chin) (shoulders) (neck) (chest) (trunk) (legs) (ankles) (feet)
16)Has your social life suffered as a result of EPP?
17)If you are over 18, do you work during the day?
18)Do you have trouble managing appointments, work, or student life on a normal schedule?
19)Do you wear special clothing prior to leaving the home?
20)Do you use any of the following on a daily basis: (sunscreen) (hat) (scarf) (face covering) (long sleeves) (gloves)
21)Do you ever purposely not use a hat, scarf or covering when out among people simply to fit in?
22)If you do neglect proper covering, how long is your recovery period until you feel back to normal?
23)Has your life improved since diagnosis?
24)Have you been more socially active since diagnosis?
25)Do you feel that knowing the name of what you are suffering from has benefitted you in any way?
Interview
The following are questions to be asked the interviewees verbally. Questions may be viewed prior to interview. The interviewer may ask follow-up questions to these if she feels that an interviewee could speak more about a certain area. All interviews will be recorded to ensure accuracy. Some questions may overlap the survey.
1)Tell me about your diagnosis how old were you and what was the process like?
2)What has been different before diagnosis and after diagnosis?
3)Were you ever isolated as a result of EPP?
4)Do you feel those situations could have been handled differently, and if so, how?
5)Do you suspect anyone else in your family has EPP?
6)How do you handle family functions or events with large groups of people in indoor/outdoor settings?
7)What has been the hardest part about having EPP?
8)In what ways has having EPP made you a better person?
9)Currently, how severe are your symptoms?
10)What is an average attack like?
11)What was the worst bout of EPP like?
12)What works for you for prevention?
13)What works for you in the case of accidental exposure?
14)If you feel pain start, what actions do you take?
15)Do you feel you need to hide your symptoms at work or with friends?
16)Do you have people you can speak openly to about your symptoms?
17)What advice would you have for an adult that suspects they may have EPP?
18) What advice would you have for a parent who recently learned their small child has EPP?
Please feel free to reach out anytime with questions or concerns.
Meghann Bauer has had symptoms of protoporphyria (EPP) since she was a little baby, yet her parents took her to different doctors for years before she was diagnosed at age 16 with EPP.
Meghann says "I just never found the right way to describe my symptoms so that the doctors would think to test me for EPP," and she was tested for lupus and many other conditions before her diagnosis. It wasn't until her mother brought Meghann to her pediatrician's office saying that she had watched Meghann fall asleep on a family car trip, and that every time the sun hit Meghann's skin she flinched away from it or woke up in pain, that doctors began to suspect porphyria.
Watching the Discovery Channel recently, Meghann found the story told in an episode of Mystery Diagnosis strikingly familiar. The show featured the Leppert family and their struggle to come up with an EPP diagnosis for their son Craig, and Meghann's family went through many of the same stages of misdiagnosis that the Leppert family did. Meghann says Craig Leppert's description of his EPP symptoms matches the feeling perfectly: "it's like putting your hand on a hot stove and then slicing your skin with a knife," a pain that is dull and steady and then suddenly sharp and searing.
Meghann has memories from her childhood of running her hands under cold water to relieve the burning pain. She and her parents did not know it was dangerous for her to be out in the sun, so she was exposed throughout the summer and the season meant constant pain. She recalls falling asleep covered in ice packs every night, and her mother describes little Meghann as having been in such pain every night, and then finally, when the ice cooled her enough and she was simply exhausted, she would heave a big sigh and go to sleep.
When she was very small, Meghann's doctors suggested she was allergic to PABA because every time her symptoms flared up she had been covered in a sunscreen containing PABA. No one realized the problem was the sun itself, not her parents' efforts to protect her.
When her pediatricians could not find anything wrong with her, they even told Meghann's parents to take her to a psychiatrist for what they assumed were psychological problems. Luckily, the psychiatrist assured Meghann's parents that there was nothing wrong with their daughter emotionally and that something physical was clearly causing her pain.
The dermatologist was surprised when tests came back positive, having said up front "you don't have EPP," something many porphyria patients have heard before. The thing about rare diseases is that while most people don't have them, some people do. And the people with those diseases are just as deserving of diagnosis and treatment as those with more common conditions. Diagnosis did not come til age 16. Yeesh!
Since she was diagnosed, Meghann takes Lumitene to help with her symptoms. She takes 10-12 pills daily throughout the summer months, and fewer in winter. Although she still has some symptoms in the summer, she is usually symptom-free all winter, except when skiing or outdoors in snow country, where the reflected sunlight causes bad symptoms. When she does have flare-ups of her EPP symptoms, she uses motrin and darvocet for pain, and packs on ice.
Meghann has also spoken with Dr. Micheline Mathews-Roth since her diagnosis, and has a genetic counselor who sees several other families in the area whose children have EPP. The genetic counselor is anxious to help and has also been in touch with Dr. Roth.
Today, Meghann is a member of the APF and a patient looking forward to participating in clinical trials of Afamelanotide from Clinuvel. She is hopeful that this may be a way to "help my husband and I do the things we enjoy, such as boating, swimming in our pool, picnics, walking our boxer puppy."
APF Meeting & Cook Family Award
Wednesday - June 21, 2017 @ 07:00:13
The American Porphyria Foundation will be hosting a Patient Education meeting for Erythropoietic Protoporphyria on Friday, July 21, 2017 in Craryville, NY at Camp Sundown. See the invite below.
Call the APF TODAY to RSVP on 1.866.APF.3635! We are looking forward to seeing you there.
CONGRATULATIONS TO THE COOK FAMILY!
The American Porphyria Foundation presents the President's Award each year to those who have assisted the APF greatly in achieving our mission to fund research, provide education, and create awareness. The awardee goes above and beyond to support their communities including patients, caregivers, families and physicians.
The President's Award 2017 has been given to the Cook Family for their outstanding service to the American Porphyria Foundation. Congratulations and thank you for all that you do. See the photo below of Caul and Cason Cook. The Cook Family: Lee Ann, Chris, Caul and Cason.
Erythropoietic protoporphyria (EPP) is characterized by cutaneous photosensitivity (usually beginning in infancy or childhood) that results in tingling, burning, pain, and itching within minutes of sun/light exposure and may be accompanied by swelling and redness. Blistering lesions are uncommon. Symptoms (which may seem out of proportion to the visible skin lesions) may persist for hours or days after the initial phototoxic reaction. Photosensitivity usually remains for life. Multiple episodes of acute photosensitivity may lead to chronic changes of sun-exposed skin (lichenification, leathery pseudovesicles, grooving around the lips) and loss of lunulae of the nails. Approximately 20%-30% of individuals with EPP have some degree of liver dysfunction, which is typically mild with slight elevations of the liver enzymes. Up to 5% may develop more advanced liver disease which may be accompanied by motor neuropathy similar to that seen in the acute porphyrias. Except for the small minority with advanced liver disease, life expectancy is not reduced.
DIAGNOSIS/TESTING:
Detection of markedly increased free erythrocyte protoporphyrin is the most sensitive and specific biochemical diagnostic test for EPP. Identification of biallelic pathogenic variants in FECH, encoding ferrochelatase, confirms the diagnosis.
MANAGEMENT:
Treatment of manifestations: There is no FDA-approved treatment for this disease or specific treatment for the acute photosensitivity. The pain is not responsive to narcotic analgesics. The only effective current treatment is prevention of the painful attacks by avoidance of sun/light (including the long-wave ultraviolet light sunlight that passes through window glass) through use of protective clothing (e.g., long sleeves, gloves, wide-brimmed hats, protective tinted glass for cars and windows). Although topical sunscreens are typically not useful, some tanning products containing creams which cause increased pigmentation may be helpful. Oral Lumitene� (β-carotene) may improve tolerance to sunlight by causing mild skin discoloration due to carotenemia. Severe liver complications are difficult to treat: cholestyramine and other porphyrin absorbents (to interrupt the enterohepatic circulation of protoporphyrin and promote its fecal excretion) and plasmapheresis and intravenous hemin are sometimes beneficial. Liver transplantation may be required. Prevention of secondary complications: Vitamin D supplementation to prevent vitamin D insufficiency due to sun avoidance. Surveillance: Monitoring of: hepatic function every 6-12 months and hepatic imaging if cholelithiasis is suspected; erythrocyte protoporphyrin levels (free and zinc-chelated), hematologic indices, and iron profile annually; vitamin D 25-OH levels. Agents/circumstances to avoid: Sunlight and UV light; for those with hepatic dysfunction, drugs that may induce cholestasis (e.g., estrogens); for those with cholestatic liver failure, use of protective filters for artificial lights in the operating room to avoid phototoxic damage. Evaluation of relatives at risk: If both FECH pathogenic variants have been identified in an affected family member, at-risk relatives can be tested as newborns or infants so that those with biallelic pathogenic variants can benefit from early intervention (sun protection) and future monitoring for signs of liver dysfunction. Therapies under investigation: Clinical trials for afamelanotide, an α-melanocyte stimulating hormone analog that increases melanin production (and thus skin pigmentation) have recently been completed.
GENETIC COUNSELING:
EPP is inherited in an autosomal recessive manner. In about 96% of cases an affected individual inherits a loss-of-function FECH allele from one parent and a low-expression FECH allele from the other parent. In about 4% of cases, an affected individual has two loss-of-function FECH alleles. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Heterozygotes (carriers) and individuals who inherit two low-expression alleles are asymptomatic. Carrier testing for at-risk family members and prenatal testing for pregnancies at increased risk are possible if the pathogenic variants in the family have been identified.
For therapy, a caring doctor had recommended I write about my illness, that I state what it has done to me and how it has affected my life and finally, what I hope will happen no matter how unrealistic. Since it took a better part of my lifetime to unravel the mysterious maladies that plagued me most of my life, it was difficult to write about it in a less compassionate form. It is important to understand when you read this that it is possible to have been born with more than one congenital anomaly. In my case, I was born with a mild Arnold Chiari Malformation which accounts for an inability to nurse, dizziness and clumsiness in infancy and adolescence. Porphyria symptoms were not present until my early to mid-teens.
I am sorry to inform you that I have never loved you. I have truly never understood you and even worse, no one else has understood you either! Starting from my earliest memories you have caused confusion and misunderstanding. You caused my parents to believe I was ill mannered at the table. Because of this I spent unspeakably cruel hours sitting at the dinner table being disciplined, eating my own regurgitated food, uncountable hours of stomach upsets, pain and tears. My mother always believed I personally rejected her since I couldn't nurse, and believed I rejected her excellent cooking efforts from the beginning of my existence. By the time I was 10, I was labeled a hypochondriac and clumsy. All this due to my dizziness, chronic mild aches and pains and chronic cramping constipation. My complaints were stonewalled. I learned to bury you. Anything was possible. I could fight you. You and my parents made me strong, the physical pain from life's normal upsets meant nothing to me. I concentrated on being the best at school, my uncontrollable mood swings hampered my social life until I conquered that too.
Occasionally throughout my adult life you would dig yourself out of your grave and I would be desperate enough to seek out Dr. Exorcist only to relive the cycle of my youth. Sometimes he would refer me to Mack the Knife, Dr. Pill Happy and Dr. Nightmare. Most of the time this proved to be a bad choice. But, this was back when I didn't realize I had a choice. I was naive enough to believe exorcism always worked and to not obey Dr. Pill Happy was unthinkable.
You taught me to dance between the rain drops. I concentrated on family, college, and a successful career. Once I passed 40, the rain storm started becoming more constant, the space between the drops became smaller and smaller. Until I reached the day when my plodding dance steps were so limited that my family could take it no longer and forcibly placed me in a wheelchair. I learned what it was like to be short and invisible. I had limited cognizance, would blank out (grand mall seizures), lose my balance, get severe flu like symptoms, severe back, shoulder, neck and all over pain and migraines, and the list goes on. I lost my business, some friends, and hold on loving life. I learned what it was like to truly wish for death. I became desperate and even more desperate while the Dr. Mack's argued over the semantics of my illness. This is where I learned that you and I had been together since birth. What a wonderful feeling to have been vindicated and to have been healed! Dr. Mack could be a good guy after all.
I cut you out of my life, but experienced the most devastating blow to discover a month after Dr. Mack's handiwork, while having a wellness celebration or farewell BBQ party about you, that you had an evil twin (according to an unsurprised Dr. Mack the Knife), yet to be named. Fortunately your twin is not identical, but is definitely a Gemini, unpredictable. He's thought to be a rarity.
I can remember the feeling of morning. A bright new day, the delicate warmth of sunshine bathing my face, the sound of birds chirping, a smell and feel of morning air through the crack in the window, the feeling of warmth and strength flowing through my body, the excitement of a whole day ahead of me to attack and enjoy life. It seems like only yesterday that I would take my nature walks with friends, challenging ourselves to exceedingly longer and longer jog-walks each day. Building a sand castle at the beach, standing in the sun watching a parade, going outdoors on a hot sunny day, an outdoor BBQ, going to a day game at the stadium, walking my dog around the block, wandering through the grocery store taking goods off the shelf, standing at the stove long enough to cook something, taking a long drive, playing chase with my little dog, the satisfaction of scrubbing a dirty rental house clean, cleaning my own house, gardening, working as I choose, doing all these things I took for granted would last my lifetime. The excitement of seeing snow on the mountains and planning a ski trip still haunts me. I will never do that again is my slogan for today. Focusing on what I can do is my personal challenge.
Now if I have slept, I wake to the feeling of burning pain everywhere I can feel my body. If I concentrate very hard I can hear a bird chirp over the loud ringing in my head, my nose is too plugged to smell my own bad breath let alone the morning breeze, and if I roll carefully off the bed I may not feel a jolt of excruciating back or leg pain. If I awake with any energy it is a blessing I savour for the few hours it may last. Sometimes I truly dread a walk from here to there. The gentle warmth of the sun has become a seering pain to me that evokes a headache, nausea, stomach pain, and more. I am almost always hot and have flashes burning up all day and night. Feeling cold is a blessing. The heat from the sun can trigger such a violent reaction that even the vision in my right eyes spasms and fails. I live with constant pain, fear of eating the wrong thing, hoping I will be able to sleep when the time comes and counting the physical things I can no longer do as a regrettably growing list. Out of the blue my heart will thump, bump and race for what seems an eternity. The head ringing often becomes loudly unbearable. I have learned to surround myself with noise to keep from going mad. Even worse is the unwanted knowledge about my body that keeps flowing in. I spent a lifetime bearing you and burying you. I spent 20 years trying to find out why these things are happening and now the why's won't stop. The list of diagnoses and pills/things I am allergic too is too long for me to remember. I usually just list the highlights on medical forms. You make me feel like a freak of nature.
Dear John, the evil twin, I want you to move out. I want to hear silence, I want to walk my dog in the sun and build a sand castle at the beach with my granddaughter. I want to hop out of bed and go grocery shopping unassisted. I want to learn to love the things I have learned to hate because of you. I want to go dancing in the evening and clean my house during the day. I want to walk ten miles on a summer day. I want to be able to eat like anyone else and not have pain, gain weight or get sick. I want to be able to toast the New Year. I don't care about my career, it is gone along with my total naivety about humanity. Most of all, I never want to have to see your friends, Dr. Exorcist, Dr. Pill Happy, Dr. Nightmare and Dr. Mack the Knife again as long as I live. I want to feel the morning!
Me
P.S: John, I have often been asked to write about you and to attempt this had caused me to be unbearable to live with for days, but a Dear John letter was a very pleasant task.
Variegate porphyria (VP) is a cutaneous porphyria (with chronic blistering skin lesions) and an acute porphyria (with severe episodic neurovisceral symptoms). The most common manifestation of VP is adult-onset cutaneous blistering lesions (subepidermal vesicles, bullae, and erosions that crust over and heal slowly) of sun-exposed skin, especially the hands and face. Other chronic skin findings include milia, scarring, thickening, and areas of decreased and increased skin pigmentation. Facial hyperpigmentation and hypertrichosis may occur. Cutaneous manifestations may improve in winter, and be less prevalent in northern regions and in dark-skinned individuals. Acute neurovisceral symptoms can occur any time after puberty, but less often in the elderly. Acute manifestations are highly variable, but may be similar from episode to episode in a patient with recurrent attacks; not all symptoms are present in a single episode; and acute symptoms may become chronic. Symptoms are more common in women than men. The most common symptoms are abdominal pain; constipation; pain in the back, chest, and extremities; anxiety; seizures; and a primarily motor neuropathy resulting in muscle weakness that may progress to quadriparesis and respiratory paralysis. Psychiatric disturbances and autonomic neuropathy can also be observed. Acute attacks may be severe and are potentially fatal.
DIAGNOSIS/TESTING:
No clinical features are specific for VP. It is important for all physicians, including primary physicians, to be aware of porphyrias and their variable features. The diagnosis is established by biochemical testing and confirmed by identification of a heterozygous pathogenic variant in PPOX, encoding the mitochondrial enzyme protoporphyrinogen oxidase (PPOX).
MANAGEMENT:
Treatment of manifestations: The first step in treating either acute neurovisceral attacks or cutaneous manifestations is to identify and remove exacerbating factors (see Agents/circumstances to avoid). Most acute neurovisceral attacks require hospital admission; the presence of seizures, motor neuropathy, and hyponatremia suggest severe disease that ideally should be managed in an ICU. Narcotic analgesics are usually required for pain. Ondansetron or a related drug can be used for nausea and vomiting; phenothiazines can be effective for nausea, agitation, and hallucinations. Although mild attacks (without seizures, weakness or hyponatremia and not requiring narcotics) can sometimes be treated in an outpatient setting with glucose loading, most attacks require treatment with intravenous hemin. Cutaneous symptoms are best managed by wearing protective clothing and avoiding exposure to sunlight. Symptoms may decrease when exacerbating factors are removed. No treatment is known to be effective in lowering porphyrin levels and reducing cutaneous symptoms. Analgesics may be needed for painful lesions and antibiotics for superimposed infection. Prevention of primary manifestations: Acute neurovisceral attacks are less likely to occur if exacerbating factors are corrected or avoided. Recurrent premenstrual acute attacks can be prevented with GnRH analogues; weekly or biweekly hemin infusions to prevent frequent noncyclical attacks may be effective, but data are lacking. Prevention of the skin manifestations requires protection from sunlight. Surveillance: Liver imaging at six-month intervals beginning at age 50 years in those who have experienced persistent elevations in porphobilinogen or porphyrins may detect early hepatocellular carcinoma. Agents/circumstances to avoid: Exacerbating factors that should be avoided include: barbiturates, sulfonamide antibiotics, griseofulvin, rifampin, most anticonvulsants including phenytoin and carbamazepine, alcohol, ergot alkaloids, metoclopramide, and progestins. Although birth control pills should generally be avoided, low-dose hormonal preparations may be tolerated. Concomitant illnesses should be treated effectively using drugs that are considered safe whenever possible. Updated lists of safe and unsafe drugs are maintained at the Web sites of the American Porphyria Foundation and the European Porphyria Network. Evaluation of relatives at risk: At-risk family members can be offered molecular genetic testing for the family-specific PPOX pathogenic variant to identify those who are heterozygous (for the purpose of counseling regarding appropriate use of drugs and avoidance of known exacerbating factors). Pregnancy management: Exacerbations during pregnancy have been treated successfully with heme arginate and heme hydroxide (hematin); while neither preparation has been studied extensively during pregnancy, limited data suggest that treatment during pregnancy is unlikely to produce adverse fetal effects.
GENETIC COUNSELING:
VP is inherited in an autosomal dominant manner with reduced penetrance. De novo pathogenic variants are rare. Each child of an individual with VP has a 50% chance of inheriting the pathogenic variant; while offspring who inherit the variant may or may not develop symptoms, most do not. Prenatal diagnosis for pregnancies at increased risk for VP is possible if the pathogenic variant in an affected family member has been identified. Of note, the presence of a PPOX pathogenic variant does not predict whether or at what age an individual will become symptomatic.
2017 International Congress of Porphyrins and Porphyria in Bordeaux, France
Thursday - June 8, 2017 @ 07:00:00
2017 International Congress of Porphyrins and Porphyria in Bordeaux, France
The International Conference is a gathering of porphyria experts and researchers from around the world and is held every two years. This conference provides them with the opportunity to share their research and discuss the present and future therapies, case studies, etc.
This event will take place at the Bordeaux Palais de la Bourse. The official Patient Day will be held on Sunday, June 25, 2017followed by Congress Days 1-4, which will conclude Wednesday, June 28, 2017.
Program for the International Congress on Porphyrins & Porphyrias Bordeaux ICPP2017
ICPP2017.ORG
Ways to Improve Your Health
Monday - June 5, 2017 @ 07:00:23
COVER SUBJECT IMPROVE YOUR HEALTH5 THINGS YOU CAN DO TODAY
Ways to Improve Your Health
00:00
13:48
WHO wants to be sick? At the very least, an illness is an inconvenience and an expense. You not only feel bad, but when you are sick, you may not be able to go to work or school, earn any money, or look after your family. You may even need someone to look after you, and you may have to pay for expensive medicines and treatment.
Well has it been said that Prevention is better than cure. Some illnesses cannot be avoided. Still, there is much you can do to slow down or even prevent the onset of illness. Consider five things that you can do today to get on the road to better health.
1 PRACTICE GOOD HYGIENE
According to the Mayo Clinic, one of the best ways to avoid getting sick and spreading illness is to wash your hands. One of the easiest ways to catch a cold or influenza is to rub your nose or your eyes when your hands have been contaminated by germs. Your best defense against such contamination is to wash your hands regularly. Good hygiene can also prevent the spread of more serious conditions, such as pneumonia and diarrheal diseases, which every year cause the death of over two million children under the age of five. Even the spread of deadly Ebola can be minimized by the simple habit of washing hands.
There are certain times when hand washing is particularly important to protect your own health and that of others. You should wash your hands:
After using the toilet.
After changing diapers or helping a child to use the toilet.
Before and after treating a wound or a cut.
Before and after being with someone who is sick.
Before preparing, serving, or eating food.
After sneezing, coughing, or blowing your nose.
After touching an animal or animal waste.
After handling garbage.
And do not take it for granted that you are cleaning your hands properly. Studies have shown that a large percentage of those who use public toilets do not wash their hands afterward or do not wash them correctly. How should you wash your hands?
Wet your hands in clean running water and apply soap.
Rub your hands together to make a lather, not forgetting to clean your nails, your thumbs, the backs of your hands, and between your fingers.
Keep rubbing for at least 20 seconds.
Rinse in clean running water.
Dry with a clean cloth or a paper towel.
Such measures are simple but can avert illness and save lives.
2 USE A SAFE WATER SUPPLY
Obtaining sufficient clean water for ones family is a regular chore in some countries. Yet, access to clean water can become a concern in any part of the world when a main supply that is usually good to drink becomes contaminated as a result of a flood, a storm, a pipe break, or some other issue. If water does not come from a safe source or is not stored correctly, it can cause parasite infestation, as well as cholera, life-threatening diarrhea, typhoid, hepatitis, and other infections. Unsafe drinking water is one of the causes of an estimated 1.7 billion cases of diarrheal disease every year.
There is much you can do to slow down or prevent the onset of illness
Cholera is most often contracted when a person drinks water or eats food that is contaminated with fecal matter from infected people. What steps can you take to protect yourself, even in the immediate aftermath of a disaster, from this and other types of water contamination?
Ensure that all your drinking waterincluding the water used for brushing teeth, making ice, washing food and dishes, or cookingcomes from a safe source, such as an adequately treated public supply or sealed bottles from a reputable firm.
If there is any possibility that your piped supply has been contaminated, boil your water before use or treat it with an appropriate chemical product.
When using chemicals, such as chlorine or water-purifying tablets, follow the makers directions carefully.
Use quality water filters, if available and affordable.
If no water-treatment products are available, add household bleach, eight drops per gallon of water (two drops per liter), mix well, and then let the water stand for 30 minutes before using it.
Always store treated water in clean, covered containers to protect it from possible recontamination.
Ensure that any vessel used to take water from your stored supply, such as a ladle, is clean.
Handle water containers with clean hands, and do not dip your hands or fingers into water used for drinking.
3 WATCH WHAT YOU EAT
Good health is impossible without good nutrition, and for good nutrition you need a healthy, balanced diet. You may need to consider your intake of salt, fats, and sugar, and you should watch your portion sizes. Include fruits and vegetables in your diet, and vary what you eat. Reading the packaging will help you to select whole-grain foods when buying bread, cereals, pasta, or rice. These are richer in nutrients and fiber than the alternatives made from refined grain. As for proteins, eat small and lean portions of meat and poultry and try to eat fish a couple of times a week, if possible. In some lands it is also possible to find protein-rich foods from vegetable sources.
If you eat too many sugars and solid fats, you risk becoming overweight. To minimize this risk, drink water instead of sweet beverages. Eat more fruit instead of sugary desserts. Limit your intake of solid fats from such items as sausages, meat, butter, cakes, cheese, and cookies. And instead of using solid fats for cooking, you may want to use healthier oils.
Too much salt, or sodium, in the diet can raise your blood pressure to an unhealthy level. If this is your problem, use the information on food packaging to keep your sodium intake low. Instead of salt, use herbs and spices to flavor your meals.
How much you eat can be as important as what you eat. So, while enjoying your food, do not keep eating after you are no longer hungry.
An issue tied to nutrition is the risk of food poisoning. Any food can poison you if it is not prepared and stored properly. Every year, 1 out of every 6 Americans falls sick from food poisoning. Most recover without lasting ill effects, but some die from it. What can you do to minimize the risk?
Vegetables grow in soil that may have been treated with manure, so wash these items carefully before preparing them.
Wash your hands, cutting board, utensils, dishes, and countertops with hot, soapy water before preparing each item.
To avoid cross-contamination, never put food on a surface or plate that was previously in contact with raw eggs, poultry, meat, or fish, without first washing that surface.
Cook until the food reaches the right temperature, and promptly refrigerate any perishable items that are not going to be eaten immediately.
Discard perishable items left at room temperature for more than two hours or one hour if air temperature exceeds 90 degrees Fahrenheit (32°C).
4 STAY PHYSICALLY ACTIVE
Regardless of your age, you need regular physical activity to stay in good shape. Many people today do not exercise enough. Why is exercise important? Staying physically active can help you to:
Sleep well.
Stay mobile.
Maintain strong bones and muscles.
Maintain or achieve a healthy weight.
Lower your risk of suffering from depression.
Lower your risk of premature death.
If you do not stay physically active, you are more likely to:
Suffer from heart disease.
Suffer from type 2 diabetes.
Develop high blood pressure.
Develop high cholesterol.
Suffer a stroke.
The kind of physical activity that is right for you depends on your age and your health, so it would be wise to consult your doctor before beginning any new exercise program. According to various recommendations, children and adolescents should get at least 60 minutes of moderate-to-vigorous activity every day. Adults should get 150 minutes of moderate activity or 75 minutes of vigorous activity every week.
Choose an activity that is fun. You might consider basketball, tennis, soccer, brisk walking, cycling, gardening, chopping wood, swimming, canoeing, jogging, or other aerobic exercise. How can you tell whether an activity is moderate or vigorous? A general guide would be that moderate activity makes you sweat, but more vigorous exercise makes it hard for you to hold a conversation while doing it.
5 GET ENOUGH SLEEP
The amount of sleep needed varies from person to person. Most newborns sleep for 16 to 18 hours a day, toddlers about 14 hours, and preschoolers about 11 or 12. School-age children generally need at least 10 hours of sleep, adolescents perhaps 9 or 10, and adults from 7 to 8.
Getting the right amount of rest should not be considered optional. According to experts, sufficient sleep is important for:
Growth and development in children and teenagers.
Learning and retention of new information.
Maintaining the right balance of hormones that impact metabolism and weight.
Cardiovascular health.
Disease prevention.
Insufficient sleep has been linked to obesity, depression, heart disease, diabetes, and tragic accidents. Surely these give us good reason to want to get enough rest.
So, what can you do if you realize that you have a problem getting enough sleep?
Try to go to bed and get up at the same time every day.
Make your bedroom quiet, dark, relaxing, and neither too warm nor too cold.
Do not watch TV or use gadgets while in bed.
Make your bed as comfortable as possible.
Avoid heavy meals, caffeine, and alcohol before bedtime.
If after applying these suggestions you still suffer from insomnia or other sleep disorderssuch as excessive daytime sleepiness or gasping for breath while sleepingyou may want to consult a qualified health-care professional.
Let's Show the FDA the Future of EPP.Our Children!
Friday - June 2, 2017 @ 07:00:11
Let's Show the FDA the Future of EPP.Our Children!
The APF is implementing a new photo campaign to the FDA. We want them to hear our voices loud and clear and see that we are still here, still struggling, and need immediate approval of Afamelanotide 16mg (Scenesse). The children are our future - let's tell our story to the FDA through photos and notes from all with EPP that this rare genetic disease has an enormous impact on our daily lives, and has since childhood! We want them to visualize and hear how you feel and how you would function with a treatment. EPP doesn't only affect you when you are having a reaction - it affects everything you do and how you must modify your life to AVOID a reaction. We will keep telling the FDA our stories until we have a treatment!
Follow the instructions below and we will handle the rest.
SEND A PHOTO by mail or email/scan of your child or you as a child. Choose a photo that shows the impact of this disease - either in full protective "gear", having a reaction, or avoiding a reaction. ** Please indicate if you would prefer your photo not be identifiable**
Write simple comments or a letter expressing your concerns and how a treatment would impact your life. (Ideas: Need approval now! Suffering every day! Want to go to school just like everyone else! It hurts like_____ (fill in blank)! Approve Afamelanotide now!)
Include your name, address, and contact information
Send photos and comments to the APF office
MAIL: American Porphyria Foundation 4900 Woodway Drive, Suite 780 Houston, Texas 77056
JESSI BRILL lives in Northern Virginia. I was diagnosed with EPP about 5 years ago, but my first episode was when I was 18 months old. As, I'm sure, many of you can relate, I ran the gauntlet growing up trying to figure out what was wrong with me. After being told that it was anything from having inactive sweat glands that caused swelling to just flat out being told it was all in my head, I stopped going to see doctors until I was 25 when I found the APF. It was after my parents were watching "Mystery Diagnosis" and saw the preview of the next episode of the little boy who was having reactions to the sun. They looked at each other after only watching the preview and said "That's Jess".
After making the 7 hr. drive to Charlotte, NC I met with Dr. Bonkovsky who tested me and all the years of no answers narrowed down to a 1 hr. meeting with him telling me that Im not crazy and there is something causing this pain and these episodes. I only have my reactions in the warmer months. I have never had a reaction during the winter. And although I love the winter, I have never been skiing or snowboarding in my life so no extended trips outside. Dr. Bonkovsky thinks that this is the reason I've never had a reaction during the winter months.
I have a 1 year old little girl (Lexi) that I pray doesn't have to go through what I did my entire life. I remember all too well what it was like growing up being the kid who couldn't go out and play. I don't want that for her. Even though what I know now would make things completely different for her, still.
So, my question for anyone willing to share for those of you with EPP who have kids, how do you do it? We went on our first family vacation this past summer and of course it was to the beach. I lasted 2 days with limited time in the sun before I couldn't do it anymore. The rest of my family was there so they took her out for me but it doesn't stop the emotions or tears from getting overwhelming when you know you can't do stuff with your child because of this. Editor's note:Porphyrias are family diseases, which affect every member of the family.
Erythropoietic Protoporphyria (EPP)
Friday - May 26, 2017 @ 15:07:03
Erythropoietic Protoporphyria (EPP)
Snapshot
Other common terms:EPP, protoporphyria, erythropoietic porphyria
Prevalence:Rare; between 1:58,000-200,000. Estimates of between 5000-10,000 globally
Causes:Inherited disease; defective enzyme causes inability to properly produce haem (heme).
Symptoms:Phototoxicity: swelling, burning, itching and redness of the skin, occurring during or after exposure to sunlight, including light passing through windows. Liver toxicity in 5% of cases. Microcytic anaemia can occur.
Treatments/cures:None proven fully effective to date. Phototoxicity can be avoided by complete avoidance of sunlight and certain artificial lights.
Erythropoietic protoporphyria (EPP) is a rare inherited metabolic disorder of the haem (heme) pathway causing severe phototoxicity (toxic reactions to light) in skin due to the build up of a phototoxic chemical in the skin.
EPP belongs to a heterogenous group of disorders (porphyrias) that result from a dysfunction of specific enzymes involved in the haem biosynthesis. Haem serves many essential functions in the body one of which is oxygen transport via haemoglobin.
Incidence
Erythropoietic Protoporphyria (EPP) reaction
Erythropoietic protoporphyria is rare, and few studies have been done on its prevalence. Geographic location generally doesnt seem to bias the incidence of EPP, although one study has suggested that the incidence in Slovenia is 1:58,000. Other studies include incidence that range from 1:75,000 to 1:200,000. Case reports suggest that EPP is prevalent globally.
It is estimated that between 5,000-10,000 individuals worldwide have EPP.
Symptoms
Typically, erythropoietic protoporphyria symptoms begin in childhood and are characterized by episodes of phototoxicity. The main symptoms are pain, which is often described as heat, prickling, itch or extreme sensitivity of skin exposed to light. The pain is often very severe, and swelling and blistering of the skin may result. Skin lesions resolve slowly often leaving waxy or pitted scars. Repeated exposure leads to scarring and waxy thickening of the skin on the backs of the knuckles and nose.
Erythropoietic Protoporphyria (EPP) reaction
As little as a few minutes of sunlight (which may overcast or window transmitted) may be sufficient to evoke symptoms. Symptoms are due to visible light (at wavelength above 400 nanometers, part of the electromagnetic spectrum). In most affected individuals, skin involvement persists throughout life, although some people become less symptomatic with time. Symptoms can be seasonal, starting early in the spring, continuing through summer and diminishing in winter. Some patients report that wind may exacerbate their symptoms.
Erythropoietic protoporphyria patients will sometimes have a mild microcytic anaemia, presumably due to the inability or reduced ability to form haem. This should not be confused with iron deficiency anaemia and patients should not take iron replacement for this as it may actually exacerbate the porphyria. Gall stones, usually pigment stones, are more common at an earlier age in EPP.
Liver failure occurs in 5% of erythropoietic protoporphyria patients; this is thought to be related to the increased work of the liver to clear the excessive intermediate by-products from the defective haem pathway. If liver failure occurs it can be fatal.
This lack of permanent symptoms in erythropoietic protoporphyria leads to frequent misdiagnosis by doctors, much to the frustration of parents and their child.
Causes
Protoporphyrin IX molecule
Erythropoietic protoporphyria is inherited and can be autosomal dominant or recessive. This disorder causes a chemical known as protoporphyrin IX to accumulate in the skin. When the skin is exposed to the sun, these molecules undergo a chemical reaction that results in swelling, severe and intolerable pain and scarring, a condition known as phototoxicity.
The pain is sometimes described as like having hot needles stuck into the skin. The lifelong pain experienced by these patients typically resigns them to become socially isolated, due to the lack of an efficacious treatment and their need to continuously avoid sunlight.
Ferrochelatase deficiency
The specific defective enzyme in erythropoietic protoporphyria is ferrochelatase, the last of eight enzymes in the porphyrin-haem pathway. Consequently there is an inability to chelate iron with protoporphyrin IX to form haem. Intermediates from the pathway accumulate before this final step and cause toxic effects which are involved in the dermal symptoms of EPP.
The gene for the ferrochelatase enzyme, which shares the same name, is located on chromosome 18q21.3. Molecular studies on the gene indicate that more than 60 different mutations exist, most of which are insertions or deletions.
Diagnosis
Diagnosis of erythropoietic protoporphyria is based on the detection of increased levels of free protoporphrin IX in red blood cells. Monitoring of liver function tests and red cell porphyrins are sometimes performed to pick up any early signs of liver failure.
Treatments
Action spectrum of PPIX
Sun avoidance by remaining indoors or wearing sun protective clothing including cotton gloves and a wide brimmed hat is the first line in erythropoietic protoporphyria management. The remaining treatment is focused upon symptomatic relief, although no effective symptomatic treatments have been reported. Patients have reported immersing affected skin in water (both hot and cold) to try and relieve the phototoxicity.
Most commercially available sunscreens are of no value as a treatment, but a large particle size titanium dioxide sunscreen may be of some benefit if used with other forms of sun protection.
Drugs such as �²-carotene, cysteine (an amino acid which has been shown to decrease photosensitivity) and cimitedine have been used with various disappointing results and because the disease is inherited, genetic counselling is recommended.
Hematin infusion may temporarily decrease the production of haem and may also result in a decrease of plasma and faecal porphyrins. Also, there is sporadic evidence that autologous blood cell transfusion with washed red blood cells may successfully induce clinical and biochemical remission for erythropoietic protoporphyria patients. Finally, narrow band UVB phototherapy as a treatment option may provide some protection, presumably through epidermal thickening and tanning.
Since sun avoidance is recommended, patients lead lives where they are in the sun for very limited time. This can prevent normal social activities and the intense pain that is experienced interferes with normal daily activities and can prevent adequate sleep.
Liver transplantation in EPP
Liver failure occurs in 2-5% of patients with erythropoietic protoporphyria; this is thought to be related to the increased workload of the liver to clear excessive intermediate by-products from the defective haem pathway. If liver failure occurs it can be fatal.
31 European liver transplants in EPP patients have been reported in medical literature from 1983 and 2008. A 2005 US study followed up 20 patients who had undergone the procedure.
When conducting transplants and other surgery in EPP patients, it has been recommended that yellow filters be fixed to surgical lights to remove light wavelengths below 470nm and prevent phototoxic reactions resulting from exposure.
Psychological and social impact
EPP has been widely recognised as having a serious impact on the quality of patients lives due to sun avoidance leading to social exclusion. Depression, anxiety due to fear of reactions and suicidal thoughts have been reported by EPP patients in the literature. A 2010 UK study has also shown that individuals with photosensitive disorders, including EPP, have a greater unemployment rate.
Misdiagnosis and invisible reaction symptoms can compound these issues as patients also report accusations of hypochondria when experiencing reactions or avoiding situations which may cause the onset of symptoms.
Prognosis
Symptoms of erythropoietic protoporphyria are usually with patients for life; however a few people have described symptoms reducing over time.
References
·Lecha, M et al., (2009). Erythropoietic protoporphyria, Orphanet Journal of Rare Diseases. 14(9) available online at http://www.ojrd.com/content/4/1/19
·Marko PB et al., (2007). Erythropoietic protoporphyria patients in Slovenia, Acta Dermatoven. 16(3):99-104.
·McGuire BM, et al, (2005). Liver transplantation for erythropoietic protoporphyria liver disease. Liver Transpl. 11(12):1590-6.
·Murphy GM, (2003). 'Diagnosis and Management of the Erythropoitetic Porphyrias', Dermatologic Therapy. 16:57-64.
·Rufener EA (1987). Erythropoietic protoporphyria: a study of its psychosocial aspects, British Journal of Dermatology. 116:703-708.
·Schneider-Yin et al., (2000). "New insights into the pathogenesis of erythropoietic protoporphyria and their impact on patient care", European Journal of Pediatrics. 125:719-725.
·Stafford R, et al., (2010). The impact of photosensitivity disorders on aspects of lifestyle. British Journal of Dermatology. 163(4):817-822.
·Thunell S, Harper P & Brun A, (2000). 'Porphyrins, Porphyrin Metabolism and Porphyrias, IV, Pathophysiology of Erythropoietic Protoporphyria - Dianosis, Care and Monitoring of the Patient', Scandinavian Journal of Clinical and Laboratory Investigation. 60:581-604.
·Todd DJ. 'Clinical Implications of the Molecular Biology of Erythropoietic Protoporphyria', Journal of European Academy of Dermatology Venereology. 11:207-213.
·Wahlin S, et al, (2008). Protection from phototoxic injury during surgery and endoscopy in erythropoietic protoporphyria.� Liver Transpl. 14(9):1340-6.
·Wahlin S, et al, (2011). Liver transplantation for erythropoietic protoporphyria in Europe.� Liver Transpl. epublished May 20.
Erythropoietic protoporphyria associations and online resources
As you know, the diagnosis of EPP is often delayed for many years. Researchers at the Icahn School of Medicine at Mount Sinai are conducting a survey study to learn more about the delayed diagnosis of EPP and how to better educate providers. This is a short survey (~5 min) for adults with EPP, or parents of children with EPP. If you would like to learn more and participate, click on the link below.
Comprehensive Porphyria Diagnostic and Treatment Center
Icahn School of Medicine at Mount Sinai
Department of Genetics and Genomic Sciences
"Remember.Research is the key to your cure!"
NORD! Learn the facts
Thursday - May 25, 2017 @ 11:54:31
NORD's Rare Disease Database provides brief introductions for patients and their families to more than 1,200 rare diseases. This is not a comprehensive database since there are nearly 7,000 diseases considered rare in the U.S. We add new topics as we are able to do so, with the help of rare disease medical experts.
If you are seeking information about a rare disease that is not in this database, we would suggest contacting the Genetic and Rare Diseases Information Center (GARD) at the National Institutes of Health. NIH has the most complete database of rare diseases in the U.S.
Representatives of patient organizations whose medical advisors are interested in assisting NORD in creating a report on a disease not currently covered in this database may write to orphan@rarediseases.org.
As you know, the diagnosis of EPP is often delayed for many years. Researchers at the Icahn School of Medicine at Mount Sinai are conducting a survey study to learn more about the delayed diagnosis of EPP and how to better educate providers. This is a short survey (~5 min) for adults with EPP, or parents of children with EPP. If you would like to learn more and participate, click on the link below.
Comprehensive Porphyria Diagnostic and Treatment Center
Icahn School of Medicine at Mount Sinai
Department of Genetics and Genomic Sciences
"Remember.Research is the key to your cure!"
Andreea Miller and her Struggle with AIP
Friday - May 19, 2017 @ 07:00:02
Andreea Miller
Type of Porphyria:
Acute Intermittent Porphyria (AIP)
I started getting symptoms in 2010 after my 19th birthday. Every few months, I would get the flu, but it was worse than the flu, because it would knock me out to the point that I couldnt get out of bed without help. My muscles and bones would not cooperate. This went on for about 2 years. It would always start with a bellyache and what seemed like heartburn. I always felt lethargic, and my bones would hurt. I needed someone to physically force me out of bed. Only hot baths would help, but it would end up being too hot and would worsen my stomach pain. I went to a few urgent care clinics but nothing popped up.
Around the middle of 2011, I began going to both of the two ERs in my town. They would do a full work-up, including blood work, CT scans, EKG, etc. Upon finding nothing, I would then head to the other ER. At one point I was told I had an ovarian cyst and removal would cure my symptoms. I agreed with everything they said. After removal, I was in even more pain than before. Hydrocodone would make me sick when they would give it to me for pain, so I began to refuse it.
In mid-2011, I started to get sick more often. I began experiencing symptoms more and more frequently. I would constantly go from one ER to the other, and they would all tell me nothing was wrong. This continued for about 6 months. Beginning in 2012, it got worse and worse. Finally, the doctors were convinced it was in my head and said I couldnt keep coming back and forth to the ER if nothing was wrong. They said they needed to perform a psychological evaluation or send me to another place because I was becoming an issue at the hospital by taking away from other patients time. My mom fought back and said, What 20 year old WANTS to be sick this badly?
My condition deteriorated and by mid-2012, I was having monthly attacks. In September 2012, my grandmother passed away and that was the final straw. We were very close, as she was the one that raised me. About a week before she passed, I was in a walker, at my own request. I had been having spinal taps, more blood work, and many other tests, so I was not able to spend much time with my grandmother in her last days. Many specialists visited me from all over town, but nothing popped up. Many suggested dehydration as the cause of my symptoms.
I had a nervous breakdown when my grandmother passed away. I went to the ER in early October 2012, a few weeks after the burial. I had lost so much hope but told my mom I thought we should try the ER one more time. While we were there, one of the doctors decided that gallstones were causing my symptoms. They operated immediately and removed my gallbladder. I was released on October 10, 2012. The next day, after leaving the hospital, everything became too overwhelming for me to handle. I went back on October 12, 2012 and called my surgeon to see if my symptoms were possibly the result of surgical complications. He told me to just take more pain medication and that they would not admit me. The last thing I remember is walking into the ER; the staff was well familiar with me at this point. I was admitted for dehydration, malnutrition, etc. While I was in my room I asked my husband to help me to the restroom, I remember my heartbeat in my head, the walls turning black around me and me telling my husband I was going to pass out. I had 3 seizures over a few hours and I had no memory of any of the next three days after that. Supposedly, I drifted in and out, but I remembered nothing. When I woke up, I couldnt speak to my mom. It felt like my tongue was stuck and I couldnt move anything. I began choking on my spit, because I had become paralyzed. Later I realized I was having neurological symptoms as a result of not being treated for porphyria. When they all realized I was not faking it anymore they began calling other doctors for a consultation. They called the Texas Tech unit and by that time, I was paralyzed from the mouth down.
Starting on October 12th, I spent 62 consecutive days in hospital without leaving. Dr. Urban came in from Texas Tech. He pulled up a chair and asked my mom if I was having my cycle and when she said no, he concluded that dehydration was not my problem. He said he had a friend that was a blood specialist in NY. He sent my blood to another specialist. A few days later he told me he knew what I had. I was ecstatic. We wanted something manageable, fixable, curable, etc. He sat us down and said, You have Acute Intermittent Porphyria and explained it all. He said its not curable, however, there are treatments. He said he was not a specialist, but had witnessed this before. I remained paralyzed for a while, but slowly regained functions such as swallowing, which returned first, and then eventually all speaking abilities.
I am still in a wheelchair because of this experience. I have attacks monthly, sometimes even twice a month. I attend physical therapy, and I would be walking if I didnt have regular attacks that practically erase any progress I have made over the last month. If I had a break from the attacks, I would be able to walk again. I mainly just experience drop foot at this point.
I always get Panhematin for attacks in my Lubbock, TX hospital. Weekly infusions would help tremendously with attacks, but since I cannot afford the treatment, the doctors apologetically told me I will have to wait until Im in an attack to get my hemin. I have had roughly 85 hospital admissions in my lifetime. I have lost count of the exact number. For over a year and a half, I spent up to 30 consecutive hours going between ERs before I received my diagnosis.
I flew to see Dr. Anderson for the first time in August 2014. I flew to UTMB to get Panhematin and establish a relationship with a porphyria specialist. While I was there, he asked me to be part of a research trial and Dr. Ede explained what the benefits there are to joining the research. I am now enrolled in every possible study that I qualify for and have no regrets about joining the studies.
To learn more about signing up for research studies in your area please visit:
Most porphyrias are inherited conditions with either an autosomal dominant or autosomal recessive pattern of inheritance. However, some forms of porphyria can be caused by environmental factors such as infections or exposures to certain prescription drugs. Porphyrias caused by environmental factors are called sporadic or acquired porphyria.[2][3]
The Human Phenotype Ontology (HPO) provides the following list of features that have been reported in people with this condition. Much of the information in the HPO comes from Orphanet, a European rare disease database. If available, the list includes a rough estimate of how common a feature is (its frequency). Frequencies are based on a specific study and may not be representative of all studies. You can use the MedlinePlus Medical Dictionary for definitions of the terms below.
Many of the signs and symptoms of porphyria are similar to those of other more common diseases. Also, because porphyria is rare, many doctors have not seen cases of the disorder before, making it more difficult to diagnosis. Because porphyria's signs and symptoms usually aren't distinctive, laboratory tests are required to make a definitive diagnosis and to determine which type of porphyria is involved.[4]If your doctor suspects porphyria, he or she may recommend the following tests:[4][5]
Urine test. If you have a form of acute porphyria, a urine test may reveal elevated levels of two substances: porphobilinogen and delta-aminolevulinic acids, as well as other porphyrins.
Blood test. If you have a form of cutaneous porphyria, a blood test may show an elevation in the level of porphyrins in the liquid part of your blood (plasma).
Stool sample test. Analysis of a stool sample may reveal elevated levels of some porphyrins that may not be detected in urine samples. This test may help your doctor determine your specific type of porphyria.
Genetic testing may also be used to confirm the diagnosis.[5]
The resources below provide information about treatment options for this condition. If you have questions about which treatment is right for you, talk to your healthcare professional.
Management Guidelines
Orphanet Emergency Guidelines is an article which is expert-authored and peer-reviewed that is intended to guide health care professionals in emergency situations involving this condition.
Research helps us better understand diseases and can lead to advances in diagnosis and treatment. This section provides resources to help you learn about medical research and ways to get involved.
Clinical Research Resources
ClinicalTrials.gov lists trials that are studying or have studied Porphyria. Click on the link to go to ClinicalTrials.gov to read descriptions of these studies.
Please note: Studies listed on the ClinicalTrials.gov website are listed for informational purposes only; being listed does not reflect an endorsement by GARD or the NIH. We strongly recommend that you talk with a trusted healthcare provider before choosing to participate in any clinical study.
Orphanet lists European clinical trials, research studies, and patient registries enrolling people with this condition.
Patient Registry
The Porphyrias Consortium is a team of doctors, nurses, research coordinators, and research labs throughout the U.S., working together to improve the lives of people with this condition through research. The Porphyrias Consortium has a registry for patients who wish to be contacted about clinical research opportunities.
Nonprofit support and advocacy groups bring together patients, families, medical professionals, and researchers. These groups often raise awareness, provide support, and develop patient-centered information. Many are the driving force behind research for better treatments and possible cures. They can direct people to research, resources, and services. Many groups also have experts who serve as medical advisors. Visit their website or contact them to learn about the services they offer. Inclusion on this list is not an endorsement by GARD.
RareConnect has an online community for patients and families with this condition so they can connect with others and share their experiences living with a rare disease. The project is a joint collaboration between EURORDIS (European Rare Disease Organisation) and NORD (National Organization for Rare Disorders).
Living with a genetic or rare disease can impact the daily lives of patients and families. These resources can help families navigate various aspects of living with a rare disease.
Financial Resources
The HealthWell Foundation provides financial assistance for underinsured patients living with chronic and life-altering conditions. They offer help with drug copayments, deductibles, and health insurance premiums for patients with specific diseases. The disease fund status can change over time, so you may need to check back if funds are not currently available.
These resources provide more information about this condition or associated symptoms. The in-depth resources contain medical and scientific language that may be hard to understand. You may want to review these resources with a medical professional.
Where to Start
Genetics Home Reference (GHR) contains information on Porphyria. This website is maintained by the National Library of Medicine.
MayoClinic.com provides information about porphyria. Click on the link to access this information.
MedlinePlus was designed by the National Library of Medicine to help you research your health questions, and it provides more information about this topic.
The National Digestive Diseases Information Clearinghouse (NDDIC), part of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), offers information on this condition. Click on the link to view information on this topic.
The National Human Genome Research Institute's (NHGRI) website has an information page on this topic. NHGRI is part of the National Institutes of Health and supports research on the structure and function of the human genome and its role in health and disease.
The National Organization for Rare Disorders (NORD) has a report for patients and families about this condition. NORD is a patient advocacy organization for individuals with rare diseases and the organizations that serve them.
In-Depth Information
Medscape Reference provides information on this topic. You may need to register to view the medical textbook, but registration is free.
The Monarch Initiative brings together data about this condition from humans and other species to help physicians and biomedical researchers. Monarchs tools are designed to make it easier to compare the signs and symptoms (phenotypes) of different diseases and discover common features. This initiative is a collaboration between several academic institutions across the world and is funded by the National Institutes of Health. Visit the website to explore the biology of this condition.
The Online Mendelian Inheritance in Man (OMIM) is a catalog of human genes and genetic disorders. Each entry has a summary of related medical articles. It is meant for health care professionals and researchers. OMIM is maintained by Johns Hopkins University School of Medicine.
Orphanet is a European reference portal for information on rare diseases and orphan drugs. Access to this database is free of charge.
PubMed is a searchable database of medical literature and lists journal articles that discuss Porphyria. Click on the link to view a sample search on this topic.
Questions sent to GARD may be posted here if the information could be helpful to others. We remove all identifying information when posting a question to protect your privacy. If you do not want your question posted, please let us know. Submit a new question
I would like to be tested for porphyria. How is this condition diagnosed? Is genetic testing available? See answer
What can you tell me about acute intermittent porphyria and Chester porphyria? See answer
We, at the American Porphyria Foundation, would like to THANK YOU for participating in National Porphyria Awareness Week. We are very grateful for everyone who submitted their photos and notified us of their awareness activities. If you are still interested in continuing to RAISE PORPHYRIA AWARENESS,contact the APF.
If you have not shared your photos with the APF, please do so, we would love to see them. Don't stop now! Continue to RAISE PORPHYRIA AWARENESS!
The challenge of living with porphyria starts with how little is known about it among friends, family and the medical community. Here are some suggestions for ways that you can help:
*SHARE knowledge about porphyria at your doctor's offices and local hospitals. You might suggest that they host a seminar or grand rounds on porphyria; ask if there is a local meeting where you can hand out materials.
*ASSIST at medical conventions or health fairs to educate family, friends, and physicians on porphyria.
*Tell your story to local media. Television, newspapers and community magazines are looking for human interest stories about people who have encountered a major illness and have undertaken the challenge.
*VOLUNTEER your talents or skills to help achieve the educational programs of the APF. For example, donate one of your paintings, sculpture, computer expertise, business acumen etc. for our fall raffle or to help APF.
*HOLD a community race, car wash or other fund raising activity.
*HONOR your loved one with a gift to the APF for a birthday, anniversary, holiday or memorial tribute.
*WRITE a letter to friends and family asking them to consider making a donation;
The APF can help you accomplish this goal by providing materials:
·Porphyria Brochures
·A Porphyria Live DVD
·Fact sheets
·A PowerPoint Presentation
·Information for Grand Rounds
·Ideas to set up Medical Seminars
·Ideas to exhibit at a Health Fair
·Information to gain press
·Doctor packets sent to your doctors
·Press Releases
Contact the APF TODAY for more information at 1.866.APF.3635!
"Remember.Research is the key to your cure!"
Darnisha Davenport's Experience with EPP
Friday - May 5, 2017 @ 07:00:00
After many years, I'm finally comfortable with being open about my health condition. This is one of the reasons that pushed me to start iKURLY. Not just for my hair, but it copes with my disorder as well. I have been living with a rare skin disorder since I was 15 years old. After so many doctor visits, dermatology consultations, and skin creams prescribed, no one could help me, not even knowing what it was. None of those things worked, just created medical bills out the roof. I stayed humble and did whatever I thought was needed to avoid the reactions, but as I get older, it's only getting worse. Recently I was diagnosed with EPP, short for Erythropoietic Protoporphyria. It is a rare skin disorder. It is not fun. It is painful, very painful and it burns. When I have outbreaks I can't do anything but step in shade, apply ice and become isolated so I can calm down. People use to make fun of me laughing at the fact me being allergic to the sun, but it's actually deeper than that. As life goes on I try to remain positive, happy because deep down inside I know I am blessed, and even though this makes me differentevery day I try to look at it as a positive, that this is just one more way God added to my uniqueness from others. It kills me sometimes though, because as a little girl all my family did was TRAVELdown South, sunny states you name it, and now when I try to go somewhere on vacation or just everyday living, I have to be cautious and stay protected. Pictured was my recent reaction. This is just a glimpse of what it looks like. My face swelled, I turn completely red, my skin was on fire..it's an excruciating pain you just have to endure until it goes away. I go through my moments. This is things people don't see or hear, I stay strong because I am strong to beat this. The tears shed, people don't know what you go through daily, they judge your strength and how you wear it. I thank God I don't look like what I've been through. Even with my disorder my goal is to continue spreading happiness and positivity. I am now a part of a Foundation that is assisting me with specialist in finding a cure, and next week I'll be starting treatments. This is my testimony. Pray for me.
To learn more about EPP or sign up for Research opportunities please click on these links:
Porphyrias are a group of metabolic disorders resulting from enzymatic defects in the heme biosynthetic pathway. Erythropoietic protoporphyria is thought to be the second most common porphyria seen in clinical practice. It is, however, commonly under-recognized and can lead to both cutaneous manifestations as well as derangement in hepatic function in a minority of patients. This review summarizes the current understanding of this disorder. Different treatment options are discussed with the goal of preventing liver damage. The roles of liver and bone marrow transplantation are also addressed.
Porphyrias are a group of metabolic disorders caused by defects in the heme biosynthetic pathway. Heme is the critical prosthetic group for numerous hemoproteins such as hemoglobin, myoglobin, catalase, and microsomal cytochromes. The term porphyria is derived from the Greek word porphyra, which means purple. The biochemical hallmark of porphyrias is overproduction and, ultimately, overexcretion of compounds called porphyrins, which have a deep red or purple color. The first clinical reports of porphyria appeared in the late nineteenth century and described a patient with severe cutaneous photosensitivity and brown pigmentation of the bones, which is characteristic of congenital erythropoietic porphyria. Soon thereafter, a case of acute porphyria was described in a person addicted to drugs who had urine the color of port wine and later died after taking the hypnotic drug sulfonomethane.
Porphyrias can present clinically either with neurovisceral symptoms and signs such as abdominal pain, constipation, and weakness or with cutaneous symptoms and signs. In hereditary coproporphyria and variegate porphyria, patients may present with both types of symptoms; in the other forms of porphyria, patients manifest only one type of clinical presentation. When considering therapy for porphyrias, it is useful to classify the disorders into two major categories: acute/inducible porphyrias or chronic cutaneous porphyrias (Table 1). Regardless of the specific type of acute porphyria or associated enzymatic defect, all of the acute porphyrias produce similar neurovisceral manifestations and should be managed in a similar manner. Management of cutaneous porphyria, though more specific to the particular type of porphyria, also involves the application of several general principles.
Erythropoietic protoporphyria (EPP) was first described by Magnus and associates in 1961.1 Bonkovsky and colleagues demonstrated that heme synthase or ferrochelatase (FECH), an inner mitochondrial enzyme, was deficient in this disorder (Figure 1).2 Since then, the gene for FECH has been localized to 18q21,3,4and approximately 120 disease-producing mutations have been described to date. The gene has 11 exons and a minimum size of 45 kb. From early studies of kindreds with EPP, most investigators concluded that the disease displayed autosomal dominant inheritance with low penetrance and an increased propensity to develop chronic liver disease,5 though autosomal recessive forms of EPP have also been described.6 The activity of ferrochelatase in clinically affected patients is approximately 1525% of normal activity. The parents of many patients are asymptomatic and phenotypically normal, though approximately one half demonstrate decreases in ferrochelatase activity. More recent familial studies have identified a single nucleotide polymorphism (IVS3-48C), which leads to a lower level of expression of the normal FECH gene allele.7 Patients develop clinical disease with less than 25% of normal FECH activity when a mutated allele is inherited along with the low-level-expressing, wild-type allele, which usually harbors the IVS3-48C polymorphism. One recent paper stressed the importance of large deletions in the FECH gene exons as an important cause of mutation-negative EPP.8
FECH carries out the ultimate step in heme biosynthesis, namely the insertion of ferrous iron into protoporphyrin for the formation of heme. The only difference between heme and protoporphyrin is the insertion of an iron atom into the tetrapyrrole ring in heme. This insertion increases the stability of the structure and results in the loss of its fluorescent properties. In EPP, the major site of protoporphyrin overproduction is localized to the bone marrow, where erythrocytes are developed.912 Protoporphyrin, unlike other porphyrins, is water-insoluble and therefore removed from the body only through hepatic excretion into bile or feces. Protoporphyrin is not excreted in urine, though it does undergo some enterohepatic circulation.
Of all the porphyrias found in the United States, EPP is thought to be second in prevalence only to porphyria cutanea tarda. The exact prevalence of EPP has not been reliably estimated due to its frequently mild nature and normal urinary porphyrins, which increase the possibility of missing bona fide cases. There is no gender predominance in EPP, which is found across all ethnic and racial groups.
EPP, a disorder with highly variable clinical expression, has been previously well characterized as a chronic cutaneous porphyria.1316 Most affected patients present in early childhood with sun sensitivity and, at times, an urticarial-type reaction. Infants and children classically develop intense burning pain from sun-exposed skin following brief exposure in the spring and summer. Several hours later, erythema, edema, and itching become prominent. Vesicles are uncommon and develop only with prolonged sun exposure. With chronic and repeated exposure, involved skin may become leathery and hyperkeratotic. This reaction is particularly prominent in a malar butterfly pattern on the face and over the knuckles of the hands. Patients usually have a chronic and stable disease course, with no identified aggravating factors.
Photoactivation of lipid-soluble protoporphyrin, which is deposited in the dermal blood vessels, leads to the characteristic skin burning from sun exposure. Chronic facial scarring, hirsutism, and fluorescent teeth, which are common in congenital erythropoietic porphyria or hepatoerythropoietic porphyria, are not usually found in EPP.16 Photoactivation of circulating water-soluble uroporphyrins, on the other hand, leads to chronic blistering, as seen in congenital erythropoietic porphyria and porphyria cutanea tarda.
The most serious complication of EPP is development of pigmentary cirrhosis of the liver.17,18 Fortunately, liver injury is evident in only a minority of patients with EPP (-10%), with approximately 35% of the patients eventually developing end-stage liver disease. The pattern of liver injury, when seen in this disorder, typically develops over a period of time and ranges from mild liver disease with elevations in liver enzymes to progressive liver disease with extensive bridging fibrosis or cirrhosis and other concomitant complications. By itself, EPP rarely presents as true acute liver injury, though there may be sudden acute worsening of underlying chronic liver disease.19 Liver disease is thought to develop due to the precipitation of protoporphyrin in hepatocytes and biliary radicles.20,21 No risk factors have been identified that are able to predict the development of liver disease in EPP thus far.
A common complication of EPP is the development of pigment gallstones, which have a high content of protoporphyrin. In bile, protoporphyrin may crystallize, providing the nidus for development of gallstones. When evident in children, gallstones should prompt evaluation for EPP.22 Mild anemia with or without hemolysis is occasionally noted. In EPP, abdominal pain and neuropathy-like symptoms are seen only rarely and in the setting of advanced liver disease.2326 Of note, motor neuropathy can develop both pre- and postoperatively at the time of liver transplantation.
The diagnosis of EPP requires the demonstration of increased levels of protoporphyrin, without increased levels of coproporphyrin, in the stool, RBCs, or both.27 In EPP, elevated levels of erythrocyte protoporphyrin, plasma protoporphyrin, and stool protoporphyrin are evident. Urinary porphyrin levels are usually normal because, as already mentioned, protoporphyrin is not excreted into the urine. Analysis of the plasma porphyrin emission spectrum reveals a characteristic peak at approximately 634636 nm, following excitation with light in the Soret band (-400 nm). Erythrocyte protoporphyrin elevations are also evident in severe iron deficiency and lead poisoning. In EPP, the protoporphyrin is primarily free protoporphyrin, in contrast to iron and lead toxicity, in which predominantly Zn-protoporphyrin is found.28In EPP patients, rising plasma protoporphyrin levels, along with severe elevations in RBC protoporphyrin levels, may portend impending liver failure.29,30 Patients with EPP who develop significant liver disease have been shown to have significant increases in urinary coproporphyrin levels. An inversion of the physiological urinary coproporphyrin isomer III/I ratio has also been observed in patients with protoporphyria who develop cholestatic cirrhosis.31 Liver biopsy is performed to assess the severity of suspected liver disease, particularly in patients with very high RBC (>5,000 µg/dL) or plasma protoporphyrin (>1,000 µg/dL) concentrations. Polarized microscopy of the biopsy may demonstrate birefringent crystal deposits with characteristic Maltese cross patterns in EPP. Plugging of biliary canaliculi with protoporphyrin may also be evident in patients with advanced liver disease.32
A multitude of treatment options are available for EPP,27 ranging from efforts to reduce protoporphyrin overproduction in the bone marrow to augmenting its excretion into bile (Table 2). Other agents are used for their cytoprotective properties in conjunction with measures to reduce the circulating pool of protoporphyrin and, on occasion, liver transplantation to correct end-organ damage. Cutaneous protection with opaque sunscreens and barrier clothing cannot be overemphasized. Standard sunscreens are not useful; the only topical sunscreens that are effective at blocking wavelengths greater than 400 nm, with a high sun protection factor (>30), are light-opaque, contain zinc oxide or titanium dioxide, and may be cosmetically unacceptable to some patients. Oral beta-carotene (Solatene, Roche; 60180 mg orally per day) reduces photosensitivity in approximately 80% of patients over a 13 month period after initiation of therapy. Beta-carotene causes yellow-orange discoloration of the skin, and topical beta-carotene cream is ineffective. Beta-carotene is thought to scavenge the free radicals generated by the excitation of protoporphyrin and is dosed to maintain a therapeutic serum level of 600800 pg/dL.33 Beta-carotene's efficacy has been questioned in the recent literature, and many patients have found their resulting yellow skin difficult to accept. Oral cysteine has also been used and is thought to have a similar mechanism of action.34 Bile-acid binding agents such as cholestyramine35,36 have been used to decrease the enterohepatic circulation of protoporphyrin. Patients have attempted to use long-term activated charcoal as a safe and cheap alternative in the hopes of decreasing intestinal reabsorption of protoporphyrin.37 Intravenous vitamin E was reported in a case report as being effective for reversing liver disease.38 Ursodeoxycholic acid has been used in patients with early liver disease,31 not only because of its ability to enhance the biliary excretion of protoporphyrin, but also because of its cytoprotective properties.3941 However, a recent study in an autosomal recessive mouse model of EPP demonstrated no benefit with the use of ursodeoxycholic acid and heme arginate.42 Patients are advised against caloric restriction and should undergo iron replacement only if they are found to be iron-deficient.4347
Chronic therapy with intravenous hematin and erythrocyte transfusions is thought to suppress heme production4851 by decreasing protoporphyrin levels. Exchange transfusions and plasmapheresis have been used to remove the protoporphyrin in transit, as a bridge to liver transplantation or other more definitive treatments. Heme therapy, along with plasmapheresis, has been noted to stabilize patients with advanced liver disease.5254 Nonbiologic liver assist devices have been used recently in an attempt to decrease the morbidity associated with motor neuropathy, both in the pre- and postoperative period.55
Since the first liver transplantation for EPP-related liver disease in 1980, EPP patients with progressive liver disease have been transplanted with acceptable outcomes. Liver transplant recipients from US centers have been shown to have 1-, 5-, and 10-year survival rates of 85%, 69%, and 47%, respectively.56 These survival rates are similar to those for liver transplants performed for other indications. However, recurrent EPP was found in 65% (11/17) of patients who survived more than 2 months post-transplant. The high incidence of recurrent disease post-transplant is not surprising, as the transplant itself does not correct the primary cause and major source of protoporphyrin overproduction, which has been shown to be in the bone marrow.57 Of all the porphyrias, EPP has the best established indication for liver transplantation as a therapeutic option.58 The previous medical literature, as well as more recent reports, have demonstrated a benefit in EPP patients who receive a bone marrow transplant.59,60 Successful bone marrow transplantation with or without liver transplantation, depending upon the severity of the liver disease, is considered the definitive treatment for EPP.
Patients with EPP are at increased risk of developing chronic liver disease and should be vaccinated for hepatitis A and B (and possibly hepatitis E in the near future) in case they are found not to be immune. They should be counseled to avoid hepatotoxins such as alcohol because of the risk of accelerating their liver disease.61 EPP patients with no evident liver disease should be monitored with liver enzymes and porphyrin levels, both serum and RBC protoporphyrin, at least every 6 months, if not more frequently, to detect early signs of liver injury.62 Patients with known chronic liver disease should also be screened regularly for hepatocellular carcinoma.
Effective management of EPP involves the judicious use of all available treatment options to prevent disease progression and possibly achieve cure. Treatment is directed at minimizing complications from sun damage, monitoring for development of liver disease, and stabilizing cholestatic liver disease once it develops, in the hopes of a more definitive treatment such as bone marrow transplantation with or without liver transplantation. The initial experience with bone marrow transplantation is promising, though the opportune moment to intervene remains unclear.
1. Magnus IA, Jarrett A, Prankerd TA, Rimington C. Erythropoietic protoporphyria. A new porphyria syndrome with solar urticaria due to protoporphyrinaemia. Lancet. 1961;2:448451. [PubMed]
2. Bonkovsky HL, Bloomer JR, Ebert PS, Mahoney MJ. Heme synthetase deficiency in human protoporphyria. Demonstration of the defect in liver and cultured skin fibroblasts. J Clin Invest. 1975;56:11391148. [PMC free article] [PubMed]
3. Taketani S, Inazawa J, Nakahashi Y, Abe T, Tokunaga R. Structure of the human ferrochelatase gene. Exon/intron gene organization and location of the gene to chromosome 18. EurJ Biochem. 1992;205:217222. [PubMed]
4. Whitcombe DM, Carter NP, Albertson DG, Smith SJ, Rhodes DA, Cox TM. Assignment of the human ferrochelatase gene (FECH) and a locus for protoporphyria to chromosome 18q22. Genomics. 1991;11:11521154. [PubMed]
5. Sarkany RP, Alexander GJ, Cox TM. Recessive inheritance of erythropoietic protoporphyria with liver failure. Lancet. 1994;343:13941396. [PubMed]
6. Gouya L, Martin-Schmitt C, Robreau AM, Austerlitz F, Da Silva V, et al. Contribution of a common single-nucleotide polymorphism to the genetic predisposition for erythropoietic protoporphyria. Am J Hum Genet. 2006;78:214. [PMC free article] [PubMed]
7. Gouya L, Puy H, Robreau AM, Bourgeois M, Lamoril J, et al. The penetrance of dominant erythropoietic protoporphyria is modulated by expression of wildtype FECH. Nat Genet. 2002;30:2728.[PubMed]
8. Whatley SD, Mason NG, Holme SA, Anstey AV, Elder GH, Badminton MN. Gene dosage analysis identifies large deletions of the FECH gene in 10% of families with erythropoietic protoporphyria. J Invest Dermatol. 2007;127:27902794. [PubMed]
9. Straka JG, Hill HD, Krikava JM, Kools AM, Bloomer JR. Immunochemical studies of ferrochelatase protein: characterization of the normal and mutant protein in bovine and human protoporphyria. Am J Hum Genet. 1991;48:7278. [PMC free article] [PubMed]
10. Rufenacht UB, Gouya L, Schneider-Yin X, Puy H, Schäfer BW, et al. Systematic analysis of molecular defects in the ferrochelatase gene from patients with erythropoietic protoporphyria. Am J Hum Genet. 1998;62:13411352. [PMC free article] [PubMed]
11. Bloomer JR, Hill HD, Kools AM, Straka JG. Heme synthesis in protoporphyria. Curr Probl Dermatol. 1991;20:135147. [PubMed]
12. Samuel D, Boboc B, Bernuau J, Bismuth H, Benhamou JP. Liver transplantation for protoporphyria. Evidence for the predominant role of the erythropoietic tissue in protoporphyrin overproduction. Gastroenterology. 1988;95:816819. [PubMed]
13. Schmidt H, Snitker G, Thomsen K, Lintrup J. Erythropoietic protoporphyria. A clinical study based on 29 cases in 14 families. Arch Dermatol. 1974;110:5864. [PubMed]
14. DeLeo VA, Poh-Fitzpatrick M, Mathews-Roth M, Harber LC. Erythropoietic protoporphyria. 10 years experience. Am J Med. 1976;60:822. [PubMed]
15. Baart de la Faille H, Bijlmer-Iest JC, van Hattum J, Koningsberger J, Rade-makers LH, van Weelden H. Erythropoietic protoporphyria: clinical aspects with emphasis on the skin. Curr Probl Dermatol. 1991;20:123134. [PubMed]
16. Holme SA, Anstey AV, Finlay AY, Elder GH, Badminton MN. Erythropoietic protoporphyria in the U.K.: clinical features and effect on quality of life. Br J Dermatol. 2006;155:574581. [PubMed]
17. Chen FP, Risheg H, Liu Y, Bloomer J. Ferrochelatase gene mutations in erythropoietic protoporphyria: focus on liver disease. Cell Mol Biol (Noisy-le-grand) 2002;48:8389. [PubMed]
18. Poh-Fitzpatrick MB. The erythropoietic porphyrias. Dermatol Clin. 1986;4:291296. [PubMed]
19. Reisenauer AK, Soon SL, Lee KK, Hanifin JM. Erythropoietic protoporphyria presenting with liver failure in adulthood. Dermatology. 2005;210:7273. [PubMed]
20. Lee RG, Avner DL, Berenson MM. Structure-function relationships of protoporphyrin-induced liver injury. Arch Pathol Lab Med. 1984;108:744746. [PubMed]
22. Todd DJ. Gallstones in children. Am J Dis Child. 1991;145:971972. [PubMed]
23. Rank JM, Carithers R, Bloomer J. Evidence for neurological dysfunction in end-stage protoporphyric liver disease. Hepatology. 1993;18:14041409. [PubMed]
24. Muley SA, Midani HA, Rank JM, Carithers R, Parry GJ. Neuropathy in erythropoietic protoporphyrias. Neurology. 1998;51:262265. [PubMed]
25. Lock G, Holstege A, Mueller AR, Christe W, Doss MO, et al. Liver failure in erythropoietic protoporphyria associated with choledocholithiasis and severe posttransplantation polyneuropathy. Liver. 1996;16:211217. [PubMed]
26. Nguyen L, Blust M, Bailin M, Melendez L, Raines DE. Photosensitivity and perioperative polyneuropathy complicating orthotopic liver transplantation in a patient with erythropoietic protoporphyria. Anesthesiology. 1999;91:11731175. [PubMed]
27. Bonkovsky HL, Thapar M. Porphyria. In: Rakel RE, Bope ET, editors. Conn's Current Therapy. Philadelphia, PA: Elsevier Health; 2008. pp. 469474.
28. Lamola AA, Piomelli S, Poh-Fitzpatrick MG, Yamane T, Harber LC. Erythropoietic protoporphyria and lead intoxication: the molecular basis for difference in cutaneous photosensitivity. II. Different binding of erythrocyte protoporphyrin to hemoglobin. J Clin Invest. 1975;56:15281535. [PMC free article][PubMed]
29. Poh-Fitzpatrick MB. Protoporphyrin metabolic balance in human protoporphyria. Gastroenterology. 1985;88:12391242. [PubMed]
30. Bloomer JR. The liver in protoporphyria. Hepatology. 1988;8:402407. [PubMed]
31. Gross U, Frank M, Doss MO. Hepatic complications of erythropoietic protoporphyria. Photodermatol Photoimmunol Photomed. 1998;14:5257. [PubMed]
32. MacDonald DM, Germain D, Perrot H. The histopathology and ultrastructure of liver disease in erythropoietic protoporphyria. Br J Dermatol. 1981;104:717. [PubMed]
33. Mathews-Roth MM, Pathak MA, Fitzpatrick TB, Harber LH, Kass EH. Beta carotene therapy for erythropoietic protoporphyria and other photosensitivity diseases. Arch Dermatol. 1977;113:12291232.[PubMed]
34. Mathews-Roth MM, Rosner B. Long-term treatment of erythropoietic protoporphyria with cysteine. Photodermatol Photoimmunol Photomed. 2002;18:307309. [PubMed]
35. McCullough AJ, Barron D, Mullen KD, Petrelli M, Park MC, et al. Fecal protoporphyrin excretion in erythropoietic protoporphyria: effect of cholestyramine and bile acid feeding. Gastroenterology. 1988;94:177181. [PubMed]
36. Bloomer JR. Pathogenesis and therapy of liver disease in protoporphyria. Yale J Biol Med. 1979;52:3948. [PMC free article] [PubMed]
37. Gorchein A, Foster GR. Liver failure in protoporphyria: long-term treatment with oral charcoal. Hepatology. 1999;29:995996. [PubMed]
38. Komatsu H, Ishii K, Imamura K, Maruyama K, Yonei Y, et al. A case of erythropoietic protoporphyria with liver cirrhosis suggesting a therapeutic value of supplementation with alpha-tocopherol. Hepatol Res. 2000;18:298309. [PubMed]
39. Pirlich M, Lochs H, Schmidt HH. Liver cirrhosis in erythropoietic protoporphyria: improvement of liver function with ursodeoxycholic acid. Am J Gastroenterol. 2001;96:34683469. [PubMed]
40. Rademakers LH, Cleton MI, Kooijman C, Baart de la Faille H, van Hattum J. Early involvement of hepatic parenchymal cells in erythrohepatic protoporphyria? An ultrastructural study of patients with and without overt liver disease and the effect of chenodeoxycholic acid treatment. Hepatology. 1990;11:449457. [PubMed]
41. Van Hattum J, Baart de la Faille H, Van den Berg JW, Edixhoven-Bosdijk A, Wilson JH. Chenodeoxycholic acid therapy in erythrohepatic protoporphyria. J Hepatol. 1986;3:407412. [PubMed]
43. Holme SA, Worwood M, Anstey AV, Elder GH, Badminton MN. Erythropoiesis and iron metabolism in dominant erythropoietic protoporphyria. Blood. 2007;110:41084110. [PubMed]
44. Holme SA, Thomas CL, Whatley SD, Bentley DP, Anstey AV, Badminton MN. Symptomatic response of erythropoietic protoporphyria to iron supplementation. J Am Acad Dermatol. 2007;56:10701072.[PubMed]
45. Gordeuk VR, Brittenham GM, Hawkins CW, Mukhtar H, Bickers DR. Iron therapy for hepatic dysfunction in erythropoietic protoporphyria. Ann Intern Med. 1986;105:2731. [PubMed]
46. Milligan A, Graham-Brown RA, Sarkany I, Baker H. Erythropoietic protoporphyria exacerbated by oral iron therapy. Br J Dermatol. 1988;119:6366. [PubMed]
47. McClements BM, Bingham A, Callender ME, Trimble ER. Erythropoietic protoporphyria and iron therapy. Br J Dermatol. 1990;122:423424. [PubMed]
48. van Wijk HJ, van Hattum J, Baart de la Faille H, van den Berg JW, Edixhoven-Bosdijk A, Wilson JH. Blood exchange and transfusion therapy for acute cholestasis in protoporphyria. Dig Dis Sci. 1988;33:16211625. [PubMed]
49. Bloomer JR, Pierach CA. Effect of hematin administration to patients with protoporphyria and liver disease. Hepatology. 1982;2:817821. [PubMed]
50. Todd DJ, Callender ME, Mayne EE, Walsh M, Burrows D. Erythropoietic protoporphyria, transfusion therapy and liver disease. Br J Dermatol. 1992;127:534537. [PubMed]
51. Lamon JM, Poh-Fitzpatrick MB, Lamola AA. Hepatic protoporphyrin production in human protoporphyria. Effects of intravenous hematin and analysis of erythrocyte protoporphyrin distribution. Gastroenterology. 1980;79:115125. [PubMed]
52. Reichheld JH, Katz E, Banner BF, Szymanski IO, Saltzman JR, Bonkovsky HL. The value of intravenous heme-albumin and plasmapheresis in reducing postoperative complications of orthotopic liver transplantation for erythropoietic protoporphyria. Transplantation. 1999;67:922928. [PubMed]
53. Dellon ES, Szczepiorkowski ZM, Dzik WH, Graeme-Cook F, Ades A, et al. Treatment of recurrent allograft dysfunction with intravenous hematin after liver transplantation for erythropoietic protoporphyria. Transplantation. 2002;73:911915. [PubMed]
54. Do KD, Banner BF, Katz E, Szymanski IO, Bonkovsky HL. Benefits of chronic plasmapheresis and intravenous heme-albumin in erythropoietic protoporphyria after orthotopic liver transplantation. Transplantation. 2002;73:469472. [PubMed]
55. Eefsen M, Rasmussen A, Wulf HC, Brock A, Hansen BA. Erythropoietic protoporphyria and pretransplantation treatment with nonbiological liver assist devices. Liver Transpl. 2007;13:655657.[PubMed]
57. Poh-Fitzpatrick MB, Wang X, Anderson KE, Bloomer JR, Bolwell B, Lichtin AE. Erythropoietic protoporphyria: altered phenotype after bone marrow transplantation for myelogenous leukemia in a patient heteroallelic for ferrochelatase gene mutations. J Am Acad Dermatol. 2002;46:861866. [PubMed]
58. Seth AK, Badminton MN, Mirza D, Russell S, Elias E. Liver transplantation for porphyria: who, when, and how? Liver Transpl. 2007;13:12191227. [PubMed]
59. Wahlin S, Aschan J, Bjornstedt M, Broome U, Harper P. Curative bone marrow transplantation in erythropoietic protoporphyria after reversal of severe cholestasis. J Hepatol. 2007;46:174179. [PubMed]
60. Rand EB, Bunin N, Cochran W, Ruchelli E, Olthoff KM, Bloomer JR. Sequential liver and bone marrow transplantation for treatment of erythropoietic protoporphyria. Pediatrics. 2006;118:e1896e1899.[PubMed]
61. Bonkovsky HL, Schned AR. Fatal liver failure in protoporphyria. Synergism between ethanol excess and the genetic defect. Gastroenterology. 1986;90:191201. [PubMed]
62. Mathews-Roth MM. The consequences of not diagnosing erythropoietic protoporphyria. Arch Dermatol. 1980;116:407. [PubMed]
Articles from Gastroenterology & Hepatology are provided here courtesy of Millenium Medical Publishing
All About the Porphyrias http://www.porphyriafoundation.com/about-porphyria
Wednesday - April 26, 2017 @ 17:13:02
About Porphyria
Click on the bold blue text below or on a topic listed on the left side of the page to read more.
Porphyria is not a single disease but a group of at least eight disorders that differ considerably from each other. A common feature in all porphyrias is the accumulation in the body of porphyrins or porphyrin precursors. Although these are normal body chemicals, they normally do not accumulate. Precisely which of these chemicals builds up depends on the type of porphyria.
The terms porphyrin and porphyria are derived from the Greek word porphyrus, meaning purple. Urine from some porphyria patients may be reddish in color due to the presence of excess porphyrins and related substances in the urine, and the urine may darken after exposure to light.
The symptoms and treatment vary significantly from one type of Porphyria to the next.
Porphyria symptoms arise mostly from effects on either the nervous system or the skin. Effects on the nervous system occur in the acute porphyrias (AIP, ADP, HCP and VP). Proper diagnosis is often delayed because the symptoms are nonspecific. Skin manifestations can include burning, blistering and scarring of sun-exposed areas.
The porphyrias are rare diseases. Taken together, all forms of porphyria afflict fewer than 200,000 people in the United States. Based on European studies, the prevalence of the most common porphyria, porphyria cutanea tarda (PCT), is 1 in 10,000, the most common acute porphyria, acute intermittent porphyria (AlP), is about 1 in 20,000, and the most common erythropoietic porphyria, erythropoietic protoporphyria (EPP), is estimated at 1 in 50,000 to 75,000. Congenital erythropoietic porphyria (CEP) is extremely rare with prevalence estimates of 1 in 1,000,000 or less. Only 6 cases of ALAD-deficiency porphyria (ADP) are documented.
EPP is the most common porphyria in childhood, and the one associated with the longest delays in diagnosis.
This section of our website includes basic information about symptoms, diagnosis and treatment of the eight types of Porphyria. You will also find information on diet and nutrition in porphyria and on the history of the disease.
Porphyria is a group of at least eight metabolic disorders that arise as a result of a malfunction in one of the eight steps in the body's synthesis of a complex molecule called heme. Heme is essential for the transport of oxygen to cells in the body. If any step in the synthesis of heme is blocked, an intermediate chemical accumulates in the cell, resulting in oxygen depletion. Those intermediate chemicals, known as porphyrins or porphyrin precursors, are the substances of which heme is composed. There are two general categories of porphyrias, those that affect the skin and those that affect the nervous system. The former are called cutaneous porphyrias. The latter are called acute porphyrias. Because the symptoms of the various porphyrias may resemble symptoms of other disorders, diagnosis may be difficult. Each type of porphyria represents a deficiency of a specific enzyme needed for the synthesis of heme. Treatment is specific to the type of porphyria. The porphyrias are inherited conditions, but don't all follow the same mode of inheritance.
Signs & Symptoms
Acute Intermittent Porphyria
Abdominal pain is the most common complaint in acute intermittent porphyria. In addition, some of the following symptoms occur with varying frequency: pain in the arms and leg, generalized weakness, vomiting, confusion, constipation, tachycardia, fluctuating blood pressure, urinary retention, psychosis, hallucinations, and seizures. The muscle weakness may progress to respiratory paralysis, necessitating artificial respiration. Porphobilinogen is elevated during the attack but may be consistently high in some patients. Urine may exhibit a purple-red color. Unlike other forms of porphyria, sun sensitivity is not present in this type.
Variegate Porphyria
Variegate porphyria is characterized by abrasions, blisters, and erosions of the skin which are commonly seen during the second and third decade. These lesions tend to heal slowly, often leaving pigmented or slightly depressed scars. The patients experience sensitivity to light and fragility of skin exposed to the sun. Although in many patients manifestations remain limited to the skin, episodes similar to those of acute porphyria are not uncommon. Therefore, the symptoms, drugs, precautionary measures, and treatment described for acute intermittent porphyria are applicable to variegate porphyria.
Hereditary Coproporphyria
The large amounts of coproporphyrin present in hereditary coproporphyrin makes the patient sensitive to sunlight, but skin disease is rarely severe in this type of porphyria. Clinically it resembles variegate porphyria except that a larger percentage of affected individuals exhibit few symptoms. Other symptoms are similar to those listed for acute intermittent porphyria.
Protoporphyria
Protoporphyria can have mild to severe light sensitivity and burning on exposure to the sunlight. Usually, the symptoms subside in twelve to twenty-four hours and heal without significant scarring or discoloration to the skin. The skin lesions may also progress to a chronic stage persisting for weeks and healing with a superficial scar. These manifestations generally begin in childhood. They are more severe in the summer and can recur throughout life. Affected skin, at times, exhibits the fragility or blister formation seen in other photosensitizing types of porphyria. Hepatobiliary dysfunction may be associated with significant liver damage.
Porphyria Cutanea Tarda
In porphyria cutanea tarda, exposed skin shows abnormalities similar to those found in variegate porphyria. They range from slight fragility of the skin to persistent scarring and disfiguration. Due to fragility of the skin, minor trauma may induce blister formation. Areas of increased and decreased pigment content may be noted on the skin. Blistering of light exposed skin and increased hair growth, especially on the face, are also characteristic.
Congenital Erythropoietic Porphyria
This is a very rare disease with approximately 150 patients reported in the world. congenital erythropoietic porphyria is often manifested shortly after birth with dark urine and sunlight sensitivity causing blistering and skin fragility. Later, brownish, fluorescent teeth, increased hair growth, and pronounced scarring may occur. In some cases, loss of fingers and toes and cartilage from ears or nose may be noted.
ALA-D Porphyria
ALA-D porphyria symptoms usually arise from effects on the nervous system and/or the skin. Sometimes, the cause of the nervous system symptoms is not clear. Skin manifestations include burning, blistering, and scarring of the sun exposed areas. The disease usually manifests after puberty, and more commonly occurs in women. The most common symptom is severe abdominal pain. Other characteristics are nausea, vomiting, rapid heart rate, increased blood pressure, confusion and/or seizures, and the passing of ALA (delta- aminolevulinic acid) in the urine.
Causes
The porphyrias are a group of inherited disorders caused by deficiencies of enzymes needed in particular steps in the synthesis of the heme molecule. Of the seven distinct disorders that make up the porphyrias, four are transmitted as autosomal dominant traits, two are transmitted as autosomal recessive traits and one is transmitted in a more complex manner.
The following summarizes the mode of inheritance and the location of the genetic abnormality associated with each specific type of porphyria:
Chromosomes, which are present in the nucleus of human cells, carry the genetic information for each individual. Human body cells normally have 46 chromosomes. Pairs of human chromosomes are numbered from 1 through 22 and the sex chromosomes are designated X and Y. Males have one X and one Y chromosome and females have two X chromosomes. Each chromosome has a short arm designated p and a long arm designated q. Chromosomes are further sub-divided into many bands that are numbered. For example, chromosome 1p34 refers to band 34 on the short arm (p) of chromosome 1. The numbered bands specify the location of the thousands of genes that are present on each chromosome.
Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother.
Recessive genetic disorders occur when an individual inherits the same abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
All individuals carry a few abnormal genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary for the appearance of the disease. The abnormal gene can be inherited from either parent, or can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy regardless of the sex of the resulting child.
Affected Populations
The prevalence of porphyria remains unknown, but clinicians suggest that a range of 1 per 500-50,000 of population is probable. Some forms of porphyria are more common in specific populations including, for example, in Finland.
Standard Therapies
The basic defect cannot presently be treated, but significant effort is being directed toward treating the underlying mechanisms that cause symptoms. Porphyria is diagnosed through blood, urine, and stool tests.
Some people who are identified as having inherited porphyria do not have symptoms. It is important for those people, as well as for those who have symptoms, to be aware of preventive measures that may help them avoid episodes of porphyria symptoms. These preventive measures include avoidance of certain drugs and alcohol for some types of porphyria. For some people, they may also include avoidance of exposure to sunlight because sun sensitivity can occur with some types of porphyria. More specific information on these preventive measures is available from the American Porphyria Foundation (see Resources section of this report).
Some of the drugs that affected individuals may need to avoid include barbiturates, tranquilizers, birth control pills, sedatives, and alcohol.
An orphan drug formerly known as Hemin, now called Panhematin (hemin for injection), is approved for use as a treatment for various forms of porphyria and is administered intravenously. It is extremely potent, and its use typically follows a period of glucose therapy that has not produced the desired results. For information, contact the sponsor:
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. government funding, and some supported by private industry, are posted on this government website.
For information about clinical trials being conducted at the National Institutes of Health (NIH) Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Information about current research on porphyria also is available from the American Porphyria Foundation. (See contact information and web site in the Resources section of this report.)
Recombinant human porphobilinogen deaminase (rhPBGD or Porphozom) was granted orphan drug designation on September 9, 2002, for the treatment of acute intermittent porphyria attacks. This drug is being developed by Zymenex, formerly known as HemeBiotech, a Scandinavian biotechnology company. Patients are being recruited (August 2005) for a study to help determine the safety and effectiveness of Porphozom for treatment of acute attacks. The study is being conducted by Dr. Herbert Bonkovsky at the University of Connecticut in Farmington and Dr. Karl Anderson at the University of Texas in Galveston.
Several other products have been granted orphan drug designation by the FDA for the investigational treatment of the porphyrias. For information, see the FDA Orphan Products and Designations List, or contact the American Porphyria Foundation.
A clinical trial, cosponsored by the National Center for Research Resources and the University of Texas Medical Branch in Galveston, is currently recruiting up to 25 patients. The trial is designed to characterize enzyme defects in patients with known or suspected porphyria and in their family members. Some participants will be selected to participate in other research involving porphyria, and analysis of samples taken from patients and/or their families will help in the study of specific mutations.
RareConnect offers a safe patient-hosted online community for patients and caregivers affected by this rare disease. For more information, visit www.rareconnect.org.
Anderson KE. Acute Intermittent Porphyria, ALA-Dehydratase-Deficient Porphyria, Congenital Erythropoietic Porphyria, Porphyria Cutanea Tarda, Variegate Porphyria and Hereditary Coproporphyria. In: NORD Guide to Rare Disorders. Lippincott Williams & Wilkins. Philadelphia, PA. 2003:490-95.
Mathews-Roth MM. Erythropoietic Porphyria. In: NORD Guide to Rare Disorders. Lippincott Williams & Wilkins. Philadelphia, PA. 2003:495-96.
Anderson KE, Sassa S, Bishop DF, et al. Disorders of Heme Synthesis. In: Scriver CR, Beaudet AL, Sly WS, et al., eds. The Metabolic Molecular Basis of Inherited Disease. 8th ed. McGraw-Hill Companies. New York, NY; 2001:3024-62.
Beers MH, Berkow R., eds. The Merck Manual, 17th ed. Whitehouse Station, NJ: Merck Research Laboratories; 1999:185-200.
Berkow R, ed. The Merck Manual-Home Edition.2nd ed. Whitehouse Station, NJ: Merck Research Laboratories; 2003:933-36.
Menkes JH, Pine Jr JW, et al., eds. Textbook of Child Neurology. 5th ed. Williams & Wilkins. Baltimore, MD; 1995:129.
REVIEW ARTICLES
Foran SE, Abel G. Guide to the porphyrias. A historical and clinical perspective. Am J Clin Pathol. 2003;119 Suppl:S86-93.
Lecha M, Herrero C, Ozalla D. Diagnosis and treatment of the hepatic porphyrias. Dermatol Ther. 2003;16:65-72.
Murphy GM. Diagnosis and management of the erythropoietic porphyrias. Dermatol Ther. 2003;16:57-64.
Downey DC. The porphyrin pathway: the final common pathway? Med Hypotheses. 2002;59:615-21.
Nordmann Y, Puy H. Human hereditary hepatic porphyrias. Clin Chim Acta. 2002;325:17-37.
Aggarwal N, Bagga R, Sawhney H, et al. Pregnancy with acute intermittent porphyria: a case report and review of the literature. J Obstet Gynaecol Res. 2002;28:160-62.
Ahmed I. Childhood porphyrias. Mayo Clin Proc. 2002;77:825-36.
Badminton MN, Elder GH. Management of acute and cutaneous porphyrias. Int J Clin Pract. 2002;56:272-78.
Deacon AC, Elder GH. ACP Best Practice No 165: frontline tests for the investigation of suspected porphyria. J Clin Pathol. 2001;54:5000-07.
Shaw PH, Mancini AJ, McConnell JP, et al. Treatment of congenital erythropoietic porphyria in children by allogenic stem cell transplantation: a case report and review of the literature. Bone Marrow Transplant. 2001;27:101-05.
The information in NORDs Rare Disease Database is for educational purposes only and is not intended to replace the advice of a physician or other qualified medical professional.
The content of the website and databases of the National Organization for Rare Disorders (NORD) is copyrighted and may not be reproduced, copied, downloaded or disseminated, in any way, for any commercial or public purpose, without prior written authorization and approval from NORD. Individuals may print one hard copy of an individual disease for personal use, provided that content is unmodified and includes NORDs copyright.
National Organization for Rare Disorders (NORD) 55 Kenosia Ave., Danbury CT 06810 â?¢ (203)744-0100
#NPAW2017 Ryan Taal & EPP~ Let's storm the doors down!
Friday - April 28, 2017 @ 07:00:30
I have suffered from EPP since I was born. Shortly after I was born, my parents went camping and had me out in the sun. I very quickly began screaming and my parents were unable to quiet me down. Issues with being in the sun have run in my family. One of my Omas has had symptoms her whole life as well as her brother and some other relatives of hers. Because of this, my parents knew quickly that I had to be kept out of the sun. One of my brothers also showed symptoms of EPP. When he was a few years old, a doctor he had been referred too had him tested for EPP. It was decided then that since he was diagnosed with it, and I had the same symptoms that I also had it. My parents were also told they should keep us inside and out of the sun as much as possible. They instead decided to leave it up to us. We decided to try and live normal lives. My friends all played soccer so I also began playing soccer. On occasion, I would wear a long sleeve shirt or wear a hat if I was playing in goal, but for the most part, I tried to look like the other guys on my team. I wore lots of sunscreen when I played, and we had started on Beta-Carotene. I have had hundreds if not thousands of reactions growing up. Many very minor with just some minor irritation on the skin or feeling a little heat, but I have also had some huge reactions. My worst reaction was the first time I tried skiing. We did not know much about the causes, so skiing in the winter seemed like a good idea. It was overcast so there wasn't much sun. It was winter so the sun wasn't as powerful. So my family decided on a weeklong ski trip with family friends. I loved skiing and spent most of the day in my ski school learning the basics and going down the bunny hill as fast as I could. That evening I was in a lot of pain but I would put bags of ice on and sit outside in the cold air. The next day I had liked skiing so much I decided to go out again, and that night I was in even more pain. We did the same thing each night and every day I always made my family take me to my class. I am known to be a bit stubborn. By the end of the trip my face has scabbed over. Under the scabs the pain went away over the next couple days and I was back out doing things. My parents took me to the zoo and struggled as they watched other kids and parents point and stare at the kid with scabs that were breaking open all over his face but I didn't notice and still remember how much fun I had that day. After that we learned that often the UV light is magnified in overcast conditions and the snow bounces UV back up much like water on a lake. I had been in some of the worst conditions for my condition. As I got older I continued playing soccer. We learned that the type of beta-carotene I was on really was not that effective but we found another type and I had more success on that. I continued playing soccer and worked my way up to the top divisions for youth soccer in the city. I also found ways to do activities with my friends and family. For summer vacations we would often go camping in the mountains and my brother and I learned how to stay covered up so we could go on hikes or play around the campsite. I often wore long sleeve shirts or sweaters so I could hang out with my friends at school and many of them were very accepting of me despite having to avoid the sun or wear clothes that were odd for the conditions. When I was in grade 12 I gave going out to the mountains in winter another try and learned to snowboard, this time with my whole body fully covered, and since then I go out with my friends from time to time to ski or snowboard with no issues from the sun. I join my friends at their cabins on the lake in summer, often hanging out in the shade or covering up to go quad riding or dirt biking or to go shoot guns or many of the other things we get up to. They offer to take me out on the lake behind the boat as the sun is setting and I have learned to wakeboard, waterski, and wake surf. I have also gone out in the day wearing a full body wetsuit, and have gone surfing off of Canada's west coast wearing the same. I still react from time to time but not near as much as in the past. I have had the chance to live a life full of experiences, and have some great people in my life. I now compete in bobsleigh and race on one of the international circuits. I still need to be careful as the sport is technically outdoors, but it allows me to cover up easily without looking strange or out of place. I look forward to being able to access treatments in the future so I can worry less about the sun and be able to do more things I have wanted to in my life. But until then I will just keep finding new ways to experience things while dealing with my condition.
Let's storm the doors down!
In 2008, a group of patients accompanied Desiree to tell FDA about the many advantages about Scenesse (Afamelanotide). It has been more than 6 years and they are still dragging their feet, for no reason. Many of you want to reach out to Dr. Marcus for an update since we have heard nothing from our October meeting. Some of you want updates and others are demanding that the Scenesse (Afamelanotide) treatment be approved immediately. Whatever your goals are please write Dr. Marcus immediately. Becks Griff will be writing a letter to Dr. Marcus daily! Send your concerns to the address below.
Dr. Kendall Marcus Director, Division of Dermatology and Dental Products Food and Drug Administration 10903 New Hampshire Avenue White Oak Campus, WO 22/Rm. 5202 HFD-540 Silver Spring, MD 20993
National Porphyria Awareness Week continues!
Dougie Spence decided to document his story via social media to bring about awareness for this rare disease and to bring hope to those affected. He will be sharing videos daily for the remainder of the week.
Subscribe to his YouTube page to continue to watch.
Jacqueline Cory wearing an EPP T-Shirt designed by Cassie Tucker in support of awareness week! Thank you for sharing, Jacqueline!
The challenge of living with porphyria starts with how little is known about it among friends, family and the medical community. That is why National Porphyria Awareness Week is so important. It provides each of YOU with the opportunity to enhance porphyria awareness in your sphere of influence. Here are some suggestions for ways that you can help:
*SHARE knowledge about porphyria at your doctor's offices and local hospitals. You might suggest that they host a seminar or grand rounds on porphyria; ask if there is a local meeting where you can hand out materials.
*ASSIST at medical conventions or health fairs to educate family, friends, and physicians on porphyria.
*Tell your story to local media. Television, newspapers and community magazines are looking for human interest stories about people who have encountered a major illness and have undertaken the challenge.
*VOLUNTEER your talents or skills to help achieve the educational programs of the APF. For example, donate one of your paintings, sculpture, computer expertise, business acumen etc. for our fall raffle or to help APF.
*HOLD a community race, car wash or other fund raising activity.
*HONOR your loved one with a gift to the APF for a birthday, anniversary, holiday or memorial tribute.
*WRITE a letter to friends and family asking them to consider making a donation;
The APF can help you accomplish this goal by providing materials:
· Porphyria Brochures
· A Porphyria Live DVD
· Fact sheets
· A PowerPoint Presentation
· Information for Grand Rounds
· Ideas to set up Medical Seminars
· Ideas to exhibit at a Health Fair
· Information to gain press
· Doctor packets sent to your doctors
· Press Releases
Contact the APF TODAY for more information at 1.866.APF.3635!
"Remember.Research is the key to your cure!"
#NPAW2017 Kelly Story & PCT
Thursday - May 4, 2017 @ 13:07:55
Kelly Story
Porphyria Cutanea Tarda (PCT)
My name is Kelly Story and I live with my husband, Chad, and two cats in Kissimmee, FL. In July of 1999, one month after our wedding, I was out of town on business, and I noticed tiny little water blisters all over my hands. The blisters didn't itch and were not painful. Although I found them to be very odd, I didn't worry much.
Over a short period of time, my skin became extremely fragile. It seemed like almost anything would cause a scrape on the skin of my hands. Plus, the blisters got much worse. My hands looked like something out of a horror movie. I was so embarrassed all of the time, and I cried a lot. I went to a dermatologist. After two visits, he told me that he was fairly certain that I had PCT but suggested that I go to a specialist. After several humbling tests and weeks of waiting, I was officially diagnosed with PCT.
The doctor said that I would have to start phlebotomies. This meant that over a pint of blood would be taken from me at one time. Even after I began the phlebotomy treatments, the blisters moved to my arms. These blisters itched tremendously. My poor husband didn't know what was happening, because, I would wake up in the middle of the night with a frenzy of scratching and crying. The itching was unbearable. I would feel so guilty afterwards. After all, I was receiving treatment for my condition and knew that I did not have a fatal disease. There are other people in the world with so much more serious problems. But, it was still uncomfortable, and my hands and arms were just so ugly.
I was tired a lot during the six month or so period I was having the phlebotomies. The doctor said that I would most likely be temporarily anemic. This was the result of having the blood taken from me. I used to do aerobic exercises at least four times a week, but that stopped. I also got headaches quite often. Finally, I was in remission. No more cuts, no more blisters, and no more itching.
I lived for a while with the dark scars on my hands from all of the sores and blisters and several lighter ones on my arms. But now, the scars have faded tremendously. You can't see any on my arms, and the ones on my hands are very pale. It may sound vain, but I never thought I would have pretty hands again, and now I do. To this day, I still cringe if I accidentally knock my hand into something. But, I am always relieved when I look down and see no scrape or cut.
Since being diagnosed with PCT, I've taken estrogen and alcohol out of my life, and I try my best to stay out of the sun. It's hard living in Florida, but I just keep applying that good old sunscreen. I'm very blessed to have such a supportive husband. I told Chad on our first date that I was high maintenance. Boy, neither one of us knew how much!
"Remember..Research is your key to a cure!"
It's National Porphyria Awareness Week!
Thursday - April 27, 2017 @ 07:00:05
Look at what's going on!
It's National Porphyria Awareness Week!
We have been getting submissions on what people are doing to RAISE PORPHYRIA AWARENESS! See their efforts below!
Jared Ulmer (Porphyria J) was contacted by a student named Ashly from Whitehall High School in Mechanicsville, PA to share his story with Ashly's honors genetics class! Way to go Ashly!
Here are some photos from people raising porphyria awareness!
Justin Hamilton created a Pic Collage to raise CEP Awareness!
Cassie Tucker and her son Mitchell created a T-shirt campaign to raise awareness for EPP! Cassie donated a portion of the proceeds to the APF! Thank you, Cassie and Mitchell!
Kristen Wheeden and her son Brady (EPP) wearing APF gear in support of NPAW!
Terri Witter wearing APF gear in support of NPAW!
Tammy Smith wearing APF gear in support of NPAW!
Lina Rebeiz wearing APF gear in support of NPAW!
Lakeshia Johnson painted her nails purple in support of NPAW!
WOW! Thank you all for your continuing support in our National Porphyria Awareness Week efforts. You can use anything in life to promote awareness!
Think about what you can do to heighten awareness! It's not too late!
National Porphyria Awareness Weekprovides each of YOU with the opportunity to enhance porphyria awareness in your local and medical communities.
The APF can help you accomplish this goal by providing materials:
Porphyria Brochures
A Porphyria Live DVD
Fact sheets
A PowerPoint Presentation
Information for Grand Rounds
Ideas to set up Medical Seminars
Ideas to exhibit at a Health Fair
Information to gain press
Doctor packets sent to your doctors
Press Releases
Contact the APF TODAY at 1.866.APF.3635 or 713.266.9617
"Remember.Research is the key to your cure!"
Abdul Waheed Butt CEP
Wednesday - April 26, 2017 @ 06:00:37
Abdul Waheed Butt
Type of Porphyria:
Congenital Erythropoietic Porphyria (CEP)
Hello I am from Pakistan. When I was born, I was completely all right. After 2 months when my mother cut my nails, there was some water. My parents checked with a skin specialist who said that I had disease of skin, (Congenital erythropoietic PORPHERIA) Gunther disease.
Now my age is 21 , and there are no special effect on me . Doctors say to avoid sunlight and that my body is completely fine.. Only my hands and mouth are affected from the disease.
If you can help me to get the treatment, I never forget your this faith work. God Bless U.
1. What is Congenital Erythropoietic Porphyria?
Congenital erythropoietic porphyria (CEP), also called Günthers disease after the doctor who described it in 1911, is an inherited disease and the rarest of the main types of porphyria, In CEP the activity of an enzyme called uroporphyrinogen III synthase (UROS) is very low. This leads to increased production of porphyrins, called type I isomer porphyrins, from the bone marrow. These porphyrins accumulate in the body, especially in the red blood cells, and cause the problems associated with CEP.
2. How common is CEP?
CEP is extremely rare. As it is so rare, the exact number of people affected by CEP is not clear. It is estimated that about 1 in every 2 3 million people are affected by CEP. CEP can affect males and females equally, and any ethnic group.
3. What are the features of CEP?
Individuals with CEP may not have all of the features described here. Different individuals may have different severity of the disease. Usually, the disease shows itself soon after birth or in early childhood, but sometimes onset of disease is delayed until adolescence or early adulthood.
Red urine is usually the first sign noticed in newborn babies with CEP. This is due to the large amount of porphyrin passed in the urine. The intensity of the redness of the urine can vary from day to day.
The skin is very sensitive to light, especially direct sunlight or intense artificial light, such as the very bright light sometimes used to treat babies with jaundice. This causes the skin to become fragile and blister or ulcerate. This most commonly happens at sun-exposed sites, for example the backs of the hands, the face, ears and scalp. The skin may take longer to heal after injury or blistering, and become infected. Repeated blisters, wounds and ulcers can cause scarring in the skin and bald patches on the scalp.
Some individuals may develop darkening of sun-exposed skin.
Eyes may also be sensitive to bright sunlight or artificial light, which can cause ulcers and scarring of the eyes. With time some patients loose their eyelashes, which make their eyes prone to irritation from small particles of dust and fibres.
Anaemia (a low haemoglobin), which varies in severity, is another feature of CEP. Anaemia develops because porphyrin damages some red blood cells which are then removed and destroyed by an organ in the abdomen called the spleen. The symptoms of anaemia include feeling tired, short of breath following minimal exertion and looking pale. A blood test will confirm the presence of anaemia.
The spleen can gradually become bigger and cause worsening of the anaemia, and a reduction in the number of platelets (the blood cells that help to form blood clots to stop bleeding) and white cells (the blood cells that fight infections) in the blood leading to increased risk of bleeding (such as repeated nose bleeds) and infections.
Teeth are discoloured by porphyrin causing them to appear reddish brown, especially the milk teeth.
CEP can occasionally cause thinning of the bones (osteoporosis). Osteoporosis can lead to breakage of bones (fracture) following minimal injury.
Excess body hair may develop, especially on the face and backs of the hands.
4. How is CEP inherited?
The low UROS enzyme activity in CEP is due to alterations (mutations) in the UROS gene that codes this enzyme. Each individual has 2 copies of every gene, one inherited from their mother, and one from their father. To develop CEP, one has to have two copies of the mutated gene, one inherited from each parent as shown in the diagram. This form of inheritance is called autosomal recessive (autosomal because the gene concerned is not located on the sex chromosomes). Although the parents of individuals with CEP can pass on the mutated gene to one of their children, they themselves do not have the disease, because they also have one normal gene. Similarly, some brothers or sisters of the affected person may also inherit one mutated gene from one of the parents, but because they also inherit a normal gene from the other parent, they do not have CEP. Individuals who have one mutated gene and a normal gene (such as the parents, and 2 of the children in this diagram) are called carriers of the disease.
= Normal UROS gene = Mutated UROS gene
Mum unaffected
Dad unaffected
Unaffected child
Unaffected child
Affected child
Unaffected child
When both mother and father are carriers, each of their (unborn) children will have a 1 in 4 risk of having CEP and a 1 in 2 risk of being a carrier. The risk that the child of a carrier will have CEP is extremely low, because their partner is very unlikely to be a carrier (the gene mutation that causes CEP is very rare in the general population), unless he/she is a close relative. All the children of someone with CEP will be carriers but are very unlikely to have CEP.
5. How is CEP diagnosed?
CEP may be suspected in children (or rarely adults) who present with the features described above. The diagnosis is confirmed by measuring porphyrin levels in the individuals blood, urine and faeces. These samples need to be protected from light until tested. A blood sample may also be taken to look for genetic mutations.
6. Can CEP be diagnosed in pregnancy?
Testing for CEP in pregnancy is not offered routinely. However CEP can be diagnosed in pregnancy in families where there is already a child with CEP. In this situation, a test called amniocentesis is carried out at about 16 weeks of pregnancy. Alternatively, another test called chorionic villus sampling is carried out at about 12 weeks of pregnancy to collect blood cells from the placenta arising from the baby. These cells are then checked for the UROS gene mutations causing CEP.
7. Is there a cure for CEP?
Currently, the only available cure for CEP is a bone marrow transplant (BMT). This involves transplanting healthy bone marrow from another person (the donor) to that of the person with CEP (the recipient). Following successful BMT, the features of CEP such as photosensitivity and anaemia will resolve. However the scarring from previous damage to the skin is permanent.
For BMT to succeed, the bone marrow of the donor needs to be a good match with the recipient. BMT is a high-risk treatment where powerful treatments to suppress the recipients immune system are initially needed to prevent rejection. BMT is currently reserved for those severely affected individuals who have a matched bone marrow donor.
8. What other treatments are available for CEP?
The treatment of CEP is aimed at preventing scarring of skin and eyes, and treatment of the complications mentioned above. Some or all of the following measures may be needed:
Protection of exposed skin from direct sunlight is required to prevent blistering and scarring. Rigorous photoprotection should include the routine use of clothing, gloves, a broad brimmed hat, scarf, long sleeves, high collars and long trousers. Conventional sunscreens (that block ultraviolet light) are not effective in CEP where the photosensitivity is to visible light. Reflectant sunscreens formulated to reflect visible light from the skin surface are required. Tinted reflectant sunscreens are available which can be mixed to match the patients individual skin colour. Examples of reflectant sunscreen products available on prescription and from chemists include:
- Ambre Solaire lotion SPF 60
- RoC total Sunblock lotion SPF 25
- Delph lotion SPF25
- Sunsense Ultra SPF 60
- Delph lotion SPF 30
- Uvistat cream SPF 22
- E45 Sun lotion SPF25
- Ultrablock cream SPF30
- E45 Sun lotion SPF50
Curtains or blinds in the house and work place may be needed to reduce the intensity of visible light. Additionally, opaque window films may be applied to the windows of buildings and/or vehicles. It is important to confirm that any window film that you select for your vehicle is legally acceptable for driving laws within your country.
Cosmetic camouflage may be used to conceal scarring of the skin. (e.g. British Association of Skin Camouflage www.skin-camouflage.net)
The eyes should be protected from sunlight by the use of tinted, wrap-around sunglasses. Specialist care from an ophthalmologist may be needed.
Skin on light-exposed areas should be protected against minor trauma to prevent long-term scarring. This can be achieved by keeping the skin well moisturised and by wearing gloves.
Skin ulcers need to be kept clean, dressed appropriately and any infections treated with antibiotic creams or tablets to speed up the healing.
Repeated scarring of the skin, especially of the fingers, can restrict joint mobility. Regular, gentle hand exercises may help to delay or prevent this.
Advice from an occupational therapist may be needed in patients who develop restricted hand function due to scarring of the skin.
Blood transfusions may be needed to treat the anaemia. However, regular blood transfusions can result in iron overload. Treatment for iron overload involves a tablet or injection. Enlargement of the spleen may worsen the anaemia, necessitating removal of the spleen by an operation.
If thinning of the bones is detected (by x-rays and bone scans), treatment with tablets may be needed.
Good oral hygiene is important to prevent tooth decay. If opening the mouth is restricted due to scarring around the mouth, a soft childrens toothbrush or an electric toothbrush may be easier to use and cause less damage to the gums.
9. Will there be new treatments available for CEP in the future?
Research is underway to cure CEP with gene therapy. This would involve correcting the abnormality of the affected persons gene. It is realistic to anticipate significant progress with this research during the next decade.
10. Can certain medications make CEP worse?
CEP is an erythropopietic porphyria which differs from acute hepatic porphyrias that can be made worse by certain medications. CEP is not made worse by any of these medications. Therefore, unless the person is allergic to a medication for any other reason, individuals with CEP have no restrictions in taking any form of medication that their health requires.
11. What other precautions do CEP patients need to take?
If an individual with CEP is having an operation, their internal organs will become exposed to very bright lights in the operating theatre. This may result in damage to tissues of internal organs, just like the skin blistering after exposure to bright light. The surgeon should be aware of this risk, in order to minimise the amount of light exposure, for example, by using special light filters.
12. Where can I get more information about CEP?
As CEP is a very rare condition, most general practitioners will have little experience of the condition. However, dermatologists and haematologists see most people with CEP and usually ask advice from a porphyria specialist centre that exist in most European countries. If you are concerned about the likelihood of passing the condition onto your children, you may be referred to a geneticist or porphyria specialist centere for information.
Although there are a number of other sources of information, the majority of which are on the internet, they may not have been validated by porphyria specialists. Most give details about all the forms of porphyria. The content on this website is based on a consensus agreed by EPNET partners.
This Patient Information is based on the leaflet written by Dermatologist Dr R Katugampola, and includes information and experience gained from her clinical research study during which she interviewed and examined more than 20 CEP patients.
You are Rare Indeed.
"Remember.Research Is The Key to Your Cure?"
My Journey with VP by Sean Albright
Wednesday - April 26, 2017 @ 00:00:18
My porphyria story is a long and emotional one I grew up having chest pain, vomiting and skin issues. nobody knew what was going on not even myself. I moved to Melbourne Florida when I was 16 and first heard about porphyria from the TV show House. My doctor's had tested for everything else so when I asked to be tested for porphyria they were fine with it. It came back with a VP diagnosis. I almost passed away due to wrong treatment for it. I now have my porphyria fairly in control and use my experience with porphyria and love of Motorsports to raise awareness for porphyria via racing. I have met some really cool people who have been here for support. a player for my local minor league hockey team has convinced me to start playing hockey. I know a few people who have porphyria and play hockey due to it being indoors. I hope one day that those with porphyria are able to live more enjoyable lives
It took over 17 years for a proper diagnosis. I have received Panhematin at least once a month if not more. The American Porphyria Foundation has saved my life by letting me know the treatment I was getting was actually making me sicker and could kill me, the APF (American Porphyria Foundation) was also very helpful in me being able to find a new doctor who knew about porphyria and the proper treatment. When Sean is feeling well he enjoys raising awareness for the APF by iracing just take a look at his car.
Sean~Thank You for sharing a glimpse of whats its like to go through with VP.
For more information on Variegate Porphyria please visit porphyriafoundation.org #NPAW2017 "RememberResearch is the key to your Cure!"
Learn all about VP Variegate Porphyria by the NIH
Tuesday - April 25, 2017 @ 07:00:17
Other Names:
Porphyria variegate; VP; Porphyria, South African type; See More
Variegate porphyria is a form of hepatic porphyria most common in the white South African population. This autosomal dominant disorder may produce acute attacks (as in acute intermittent porphyria) as well as skin photosensitivity.[1][2] The condition is caused by mutations in the PPOXgene which lead to deficiency of the enzyme protoporphyrinogen oxidase.[3] Acute attacks are managed and may be prevented as in acute intermittent porphyria.[1]
Episodes of acute variegate porphyria can cause abdominal pain, vomiting, constipation, and diarrhea. During an episode, a person may also experience muscle weakness, seizures, fever, and mental changes such as anxiety and hallucinations. Individuals with variegate porphyria may also develop cutaneous symptoms, including skin photosensitivity.[3]
Last updated: 7/19/2010
The Human Phenotype Ontology (HPO) provides the following list of features that have been reported in people with this condition. Much of the information in the HPO comes from Orphanet, a European rare disease database. If available, the list includes a rough estimate of how common a feature is (its frequency). Frequencies are based on a specific study and may not be representative of all studies. You can use the MedlinePlus Medical Dictionary for definitions of the terms below.
Variegate porphyria is caused by mutations in the PPOXgene.[3] Mutations in the PPOX gene reduce the activity of protoporphyrinogen oxidase, allowing compounds called porphyrin precursors to build up in the body. These compounds are formed during the normal process of heme production, but reduced activity of protoporphyrinogen oxidase allows them to accumulate to toxic levels. More than 130 mutations in the PPOX gene have been identified in people with variegate porphyria. A particular PPOX gene mutation is found in about 95 percent of South African families with the disorder.[4]
Nongenetic factors such as certain drugs, alcohol, dieting, as well as other genetic factors that have not been identified, also contribute to the characteristic features of variegate porphyria.[4]
Variegate porphyria is inherited in an autosomal dominant manner, which means one copy of the gene in each cell is mutated. This single mutation is sufficient to reduce the activity of an enzyme needed for heme production, which increases the risk of developing signs and symptoms of porphyria.[1][3]
The diagnosis of variegate porphyria is made by finding excess coproporphyrin in urine and both coproporphyrin and protoporphyrin in feces.[1][2] The most sensitive screening test is probably a plasma porphyrin assay.[1]
Last updated: 7/19/2010
Testing Resources
The Genetic Testing Registry (GTR) provides information about the genetic tests for this condition. The intended audience for the GTR is health care providers and researchers. Patients and consumers with specific questions about a genetic test should contact a health care provider or a genetics professional.
Acute attacks are managed and may be prevented as in acute intermittent porphyria.[1][2] Hospitalization is often necessary for acute attacks. Medications for pain, nausea and vomiting, and close observation are generally required. A high intake of glucose or other carbohydrates can help suppress disease activity and can be given by vein or by mouth. Intravenous heme therapy is more potent in suppressing disease activity. It can be started after a trial of glucose therapy. However, the response to heme therapy is best if started early in an attack. Heme must be administered by vein. Panhematin is the only commercially available heme therapy for treatment and prevention of acute porphyric attacks in the United States. Heme arginate, which is marketed in some other countries, is another preparation of heme for intravenous administration.[5]
Last updated: 7/19/2010
Management Guidelines
Orphanet Emergency Guidelines is an article which is expert-authored and peer-reviewed that is intended to guide health care professionals in emergency situations involving this condition.
The American Porphyria Foundation offers a document that includes information about porphyria, types, testing, and treatment with Panhematin. Click the "document" link above to view these guidelines.
Research helps us better understand diseases and can lead to advances in diagnosis and treatment. This section provides resources to help you learn about medical research and ways to get involved.
Patient Registry
The Porphyrias Consortium is a team of doctors, nurses, research coordinators, and research labs throughout the U.S., working together to improve the lives of people with this condition through research. The Porphyrias Consortium has a registry for patients who wish to be contacted about clinical research opportunities.
Nonprofit support and advocacy groups bring together patients, families, medical professionals, and researchers. These groups often raise awareness, provide support, and develop patient-centered information. Many are the driving force behind research for better treatments and possible cures. They can direct people to research, resources, and services. Many groups also have experts who serve as medical advisors. Visit their website or contact them to learn about the services they offer. Inclusion on this list is not an endorsement by GARD.
RareConnect has an online community for patients and families with this condition so they can connect with others and share their experiences living with a rare disease. The project is a joint collaboration between EURORDIS (European Rare Disease Organisation) and NORD (National Organization for Rare Disorders).
Living with a genetic or rare disease can impact the daily lives of patients and families. These resources can help families navigate various aspects of living with a rare disease.
The HealthWell Foundation provides financial assistance for underinsured patients living with chronic and life-altering conditions. They offer help with drug copayments, deductibles, and health insurance premiums for patients with specific diseases. The disease fund status can change over time, so you may need to check back if funds are not currently available.
These resources provide more information about this condition or associated symptoms. The in-depth resources contain medical and scientific language that may be hard to understand. You may want to review these resources with a medical professional.
Where to Start
Genetics Home Reference (GHR) contains information on Variegate porphyria. This website is maintained by the National Library of Medicine.
MedlinePlus was designed by the National Library of Medicine to help you research your health questions, and it provides more information about this topic.
The National Digestive Diseases Information Clearinghouse (NDDIC), part of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), offers information on this condition. Click on the link to view information on this topic.
The National Organization for Rare Disorders (NORD) has a report for patients and families about this condition. NORD is a patient advocacy organization for individuals with rare diseases and the organizations that serve them.
In-Depth Information
Medscape Reference provides information on this topic. You may need to register to view the medical textbook, but registration is free.
The Monarch Initiative brings together data about this condition from humans and other species to help physicians and biomedical researchers. Monarchs tools are designed to make it easier to compare the signs and symptoms (phenotypes) of different diseases and discover common features. This initiative is a collaboration between several academic institutions across the world and is funded by the National Institutes of Health. Visit the website to explore the biology of this condition.
Online Mendelian Inheritance in Man (OMIM) is a catalog of human genes and genetic disorders. Each entry has a summary of related medical articles. It is meant for health care professionals and researchers. OMIM is maintained by Johns Hopkins University School of Medicine.
Orphanet is a European reference portal for information on rare diseases and orphan drugs. Access to this database is free of charge.
PubMed is a searchable database of medical literature and lists journal articles that discuss Variegate porphyria. Click on the link to view a sample search on this topic.
Questions sent to GARD may be posted here if the information could be helpful to others. We remove all identifying information when posting a question to protect your privacy. If you do not want your question posted, please let us know. Submit a new question
I have just had my DNA report returned to find that I am a carrier of variegate porphyia as is my daughter. We both are asymptomatic. I am not South African Dutch. How many second generation and now third generation Americans are diagnosed yearly? If I have never had symptoms (age 65), will I have symptoms?See answer
HCP is due to a mutation in coproporphyinogen oxidase (CPOX), which is part of the pathway that produces porphyrins and heme. It is an autosomal dominant disorder, meaning that a mutation is present in only one of the pair of CPOX genes. The incidence of active HCP appears to be at most 2 per 1,000,000. The prevalence of the genetic carrier state is unknown.
Who gets Hereditary Coproporphyria?
HCP is termed a disease with low penetrance, meaning that many genetic carriers (defined by having a CPOX mutation) never have signs or symptoms of active porphyria. Active disease in general requires the presence of environmental factors such as certain drugs, hormones, and dietary changes, as in AIP. Lists are available of drugs that are risky for HCP genetic carriers as well as drugs that are safe (http://www.porphyriafoundation.com/testing-and-treatment/drug-safety-in-acute-porphyria). The worst offenders are barbiturates, sulfonamide antibiotics, anti-seizure drugs, rifampin, and oral contraceptives (progesterone, in particular). Attacks in women may occur after ovulation and during the last part of the menstrual cycle when progesterone levels are high. Reduced food intake, often in an effort to lose weight, as well as infections, surgery, and stressful situations may also precipitate attacks. Alcohol has been implicated in some attacks. People with repeated attacks are at risk for developing chronic renal disease and liver cancer (hepatocellular carcinoma)
How is Hereditary Coproporphyria diagnosed?
The initial test for people with symptoms is quantitative urinary aminolevulinic acid (ALA), porphobilinogen (PBG) and porphyrins. Elevation of ALA, PBG and coproporphyrin (predominantly isomer III) is highly suggestive of HCP. For asymptomatic individuals, the urine studies may be normal, but a fecal porphyrin analysis will show elevation of coproporphyrin III. Screening tests of this kind should be confirmed by DNA analysis to confirm a CPOX mutation.
What are treatments for Hereditary Coproporphyria?
Treatment, complications, and preventive measures are the same as in AIP. Hospitalization is often necessary for acute attacks. Medications for pain, nausea, and vomiting and close observation are generally required. During treatment of an attack, attention should be given to sodium (salt) and water balance. Harmful drugs should be stopped. Attacks are treated with either glucose loading or hemin (Panhematin, Recordati). These are specific treatments that lower the production of heme pathway intermediates by the liver. Glucose or other carbohydrates are given by mouth if possible, otherwise by vein. However, unless an attack is mild, it is now common practice to give hemin as soon as it is available, because it works more quickly than glucose loading, preventing the neurological complications of prolonged attacks.
Patients with severe renal disease tolerate hemodialysis or kidney transplantation. Liver transplantation has been very effective for patients who have repeated attacks and who are resistant to other treatments. However, experience with transplantation as a treatment is still limited.
What is the long-term outlook after an attack of Hereditary Coproporphyria?
The prognosis is usually good if the disease is recognized and treated promptly, before nerve damage develops. Although symptoms usually resolve after an attack, recovery of neuromuscular function (in a severe case) may require several months or longer. Mental symptoms may occur during attacks but are not chronic. Premenstrual attacks often resolve quickly with the onset of menses.
Can attacks be prevented?
Yes, particularly with regard to drugs and diet. Genetic HCP carriers should become informed on drugs and other factors that can lead to symptoms (see above). They should be prepared to point their healthcare providers to drugs and medications to avoid. The American Porphyria Foundation offers a mobile phone app that pulls up this information on line (http://porphyriadrugs.com/). A Medic Alert bracelet is useful for a situation in which the patient is incapacitated. Very frequent premenstrual attacks can be prevented by a gonadotropin-releasing hormone (GnRH) analogue administered with expert guidance. In selected cases, frequent noncyclic attacks can be prevented by once- or twice-weekly infusions of hemin.
Individuals who are prone to attacks should consume a normal balanced diet. Despite on-line discussion, there is no evidence that pushing carbohydrate prevents attacks, and it has the side effect of weight gain, which is undesirable for most people. Fasting, fad diets (for example, high protein) and gastric reduction surgery should be avoided. If weight loss is desired, it is advisable to consult a physician and a dietitian about an individualized diet with modest caloric restriction (ca. 10%), which will produce gradual weight loss without increasing the risk of an attack of porphyria. Exercise is safe in porphyria, and recommended.
These Emergency Room Guidelines (download PDF) cover essential information for the emergency physician treating a patient in an acute porphyria attack, including common precipitating factors, typical presentation and other diagnostic clues, making the initial diagnosis, common sequelae and best practices for treatment. A PowerPoint presentation (as PDF) for instruction is also available.
Please note: These Guidelines are for Physician Use Only. The APF sells a separate, personalized Emergency Room and Primary Care Physician Kit that contains all the information acute porphyria patients need to have with them in the Emergency Room (medical journal articles, information about diagnostic labs and Panhematin, room for your own diagnostic test results).
Neville R Pimstone MD, PhD, Head Liver Diseases Greater West Los Angeles Veterans Affairs, Professor Emeritus UC Davis Karl E. Anderson MD, Professor, Departments of Preventive Medicine and Community Health, Internal Medicine, Pharmacology and Toxicology; Associate Director, General Clinical Research Center, Director, Porphyria Center and Laboratory, University of Texas Medical Branch, Galveston, Texas Bradley Freilich, MD, Kansas City Gastroenterology and Hepatology, LLC
KEY POINTS
1. The human porphyrias are clinical disorders reflecting defects in heme biosynthesis. 2. Acute porphyrias cause acute attacks of neurological symptoms that can be life-threatening. 3. Acute attacks are triggered by certain drugs, sex steroid hormones, reduced intake of calories and carbohydrate, alcohol and unknown factors. 4. Many of these factors stimulate heme synthesis in the liver, which in the face of a metabolic enzyme defect, leads to increased production of heme precursors that may be neurotoxic. 5. Delta-aminolevulinic acid (ALA) and porphobilinogen (PBG), are porphyrin precursors and intermediates in the heme biosynthetic pathway. 6. ALA and porphobilinogen (PBG) are almost always elevated in urine during an acute attack of porphyria. 7. The most common emergency room (ER) clinical presentation is acute abdominal pain. Other features may include seizures, confusion and hallucinations, and a progressive polyaxonal motor neuropathy, which can progress to paralysis and respiratory failure requiring a ventilator. 8. A high index of suspicion in the presence of nonspecific symptoms is important for diagnosis. A family history of porphyria, female sex, onset during the luteal phase of the menstrual cycle, or recent use of a porphyrinogenic drug may be diagnostic clues. 9. A new diagnosis of porphyria as the cause of acute symptoms must be substantiated by finding a substantial increase in urine porphobilinogen (PBG). 10. Treatment should start promptly after the diagnosis is made. Mild attacks are sometimes treated with glucose loading (e.g. 3L of 10% glucose daily by vein). 11. Most acute attacks should be treated with hemin (Panhematin, Recordati Rare Diseases at: www.recordatirarediseases.com or 866-654-0539) 3-4mg/kg into a large peripheral vein or venous access port daily for 4 days. Reconstituting Panhematin with human serum albumin rather than sterile water is recommended prior to infusion. This helps prevent phlebitis at the site of intravenous infusion. 12. Hospitalization is usually required for symptomatic treatment of pain, nausea and vomiting, correction of electrolyte imbalance and observation for respiratory impairment, either to a general medical service or ICU.
#NPAW2017 Marlene Brezee & AIP
Saturday - April 22, 2017 @ 07:00:13
Marlene Brezee
It's hard to believe it's been thirty years since my diagnosis. I thought life as I had known it was over. I had a loving husband, two wonderful children and nothing really bad had ever happened to me. I found out at the age of thirty that I was going to have another child. Everything went fairly well up until about five months into the pregnancy. I began to have abdominal pains. The pain would come and go. I would go into the hospital, stay a while, be diagnosed with false labor and given medication. In and out- In and out. The pain, however, became increasingly worse and was accompanied by nausea and vomiting. I also had a strange thing start happening. My urine turned tea colored.
Meanwhile, back into the hospital they were trying to discover what was wrong and gave me more medication, another consult with another doctor, and again the diagnosis of false labor.
My doctor decided to try an old remedy of using an alcohol I.V. to stop the false labor; I was now in a lot of distress and my moaning and groaning could be heard outside of my room. The other patients began asking what was wrong with that woman? Things then went from bad to worse, and I was sent by ambulance to the University Of Michigan Hospital in Ann Arbor. It was a frightening time in my life. It's very scary when you are so very ill, and the doctors can't find out why. My family and friends were praying so hard for me. My husband was such a comfort and rock when with me but told me later how he had sat out in the parking lot crying and praying when things seemed so dark.
God's timing is always perfect. It doesn't, however, mean that He answers as quickly as we would like. At the U of M Hospital they immediately took all medications away from me, including the pain meds. I now felt desperate. I begged them and my family to have them do surgery to see what was wrong. I knew if they would just go in and look, they would see the problem. The doctors had discussed a possible bowel problem but they really didn't know. As I say, God's timing was prefect and oh so merciful. In a few hours I was to have an exploratory laparoscope. At five months my baby would be at great risk.
How I remember the words from the two doctors standing by me as they looked down at my catheter. Those few words would change my life. "I don't like the color of the urine; The test is simple enough, do it;" The test came back positive for porphobilinogen and subsequent tests were positive for porphobilinogen. All surgery was now stopped and the diagnosis of Acute Intermittent Porphyria was given. My local doctor told me later, I was so close. I kept thinking, "What causes abdominal pain and red urine? " ; He couldn't remember, but you see, God knew the timing wasn't right. Perhaps we all had more to learn. As I was to find out later, Porphyria can be called the "little Imitator," as it can mimic so many other conditionsThe doctors realized that the very meds that had been given to help me were in fact making my symptoms worse.
My life as I had known it thirty years ago was not over for me. The baby, even though he was born two months prematurely, is now six feet three inches tall, married and in full time Christian work. I have told him how God protected him in his mothers womb. At times I tend to worry over him, as I do with all my children, but I like to remind myself that God loves him more than I ever could.
I cannot say that I have never been bothered with Porphyria again, but never for that long. I have had some episodes of severe nausea and vomiting especially after surgery or colonoscopies, and I did have a couple episodes two or three years ago that were suspect. The one started out with my head feeling whoozy, and I started to feel nauseous. I have migraine headaches, so I wasn't sure what was happening. I went to get up and walk and was so tired I sat right on the floor for awhile. I was able to get into the car but while getting to the doctor's office my legs and arms felt very strange and very weak. The only releif I could get was to hang my head as close to the car floor as I could get it. By the time I got to the doctor's office, I was so weak that the doctor described it as flacid. I was sent by ambulance from there to the ER. I received one IV glucose push in the doctor's office by the paramedic and one or two more at the hospital. I responded very well. Late that evening they started a 24 hour urine test.
For whatever reason the test did not show positive. This is still a puzzle to me. I had been taking a drug that my doctor felt that may have been the reason I got into trouble. She says she won't ever prescribe another similar one for me.
At age 61 looking back to the day of diagnosis, I realize I have done very well. When I feel good, which is most of the time, I feel so good that it is actually hard to imagine being sick.
National Porphyria Awareness Week: April 22 - 29, 2017
Saturday - April 22, 2017 @ 06:30:00
National Porphyria Awareness Week: April 22 - 29, 2017
Rita Ladner set up a table at her local pharmacy (Boone's Pharmacy) to pass out porphyria brochures and porphyria fact sheets. She also educated those with questions about this rare disease. She had one lady come up to her who had a friend recently pass away with porphyria. She had not heard of it again until she had spoken to Rita. She sends her inspiration for YOU to do MORE! Way to go, Rita!
Think about what you can do to heighten awareness!
National Porphyria Awareness Week provides each of YOU with the opportunity to enhance porphyria awareness in your local and medical communities.
The APF can help you accomplish this goal by providing materials:
Porphyria Brochures
A Porphyria Live DVD
Fact sheets
A PowerPoint Presentation
Information for Grand Rounds
Ideas to set up Medical Seminars
Ideas to exhibit at a Health Fair
Information to gain press
Doctor packets sent to your doctors
Press Releases
Contact the APF today 1.866.APF.3635!
"Remember.Research is the key to your cure!"
Patient Education Meeting on the beautiful emerald coast in Santa Rosa Beach, Florida on Wednesday, May 10, 2017. You are invited to attend!
Friday - April 21, 2017 @ 18:49:13
MARK YOUR CALENDARS!
The American Porphyria Foundation will be hosting a Patient Education
Meeting on the beautiful emerald coast in Santa Rosa Beach, Florida on Wednesday, May 10, 2017. You are invited to attend!
Family and Friends are also welcome to come. See the invite below.
Are you interested in hosting a patient education meeting in your area? Call the APF TODAY to find out more information!
1.866.APF.3635 or 713.266.9617
"Remember.Research is the key to your cure!"
National Porphyria Awareness Week: April 22 - 29, 2017
Wednesday - April 19, 2017 @ 20:10:34
NPAW 2017
Let's Raise Porphyria Awareness!
National Porphyria Awareness Week: April 22 - 29, 2017
Jake Velasquez will be hosting an APF fundraiser to raise awareness for porphyria in the New York City area. Stay tuned for details!
Candace Colbert hand distributed a number of brochures and EPP fact sheets to local hospitals and physician clinics in her area.
Think about what you can do to heighten awareness!
National Porphyria Awareness Week provides each of YOU with the opportunity to enhance porphyria awareness in your local and medical communities.
The APF can help you accomplish this goal by providing materials:
Porphyria Brochures
A Porphyria Live DVD
Fact sheets
A PowerPoint Presentation
Information for Grand Rounds
Ideas to set up Medical Seminars
Ideas to exhibit at a Health Fair
Information to gain press
Doctor packets sent to your doctors
Press Releases
Contact the APF today to get involved at 1.866.APF.3635 !
"Remember.Research is the key to your cure!"
Story~ ER DR Who Told Me I Was "Wasting Time and Resources"
Wednesday - April 19, 2017 @ 20:19:59
To the ER Doctor Who Told Me I Was 'Wasting Time and Resources,' Thank You!2
Dear Emergency Room Doctor,
Thank you for being such a jerk to me!
You may not have realized when you said I was wasting emergency department time and resources over and over that you changed me, but you did. While you may have thought I had just a headache and was taking up space for patients in the waiting area who needed to be seen, you were wrong.
See, I heard how it started. Its just a curtain after all. I heard the triage nurse slam my file down on the desk and say the person in my room was here for a headache. I know how it must have seemed when you had a full ER for the entire three hours I sat out there waiting to be seen. You didnt get it because you didnt listen.
Listening is a key part of2 your job. Its a part you failed at that day. You and the triage nurse decided I was there for drugs. I get that. Im sure you get that a lot. But you missed that I had been in the hospital for three days and was just released a few days prior. You missed that I said the hospital neurologist on call had already called to say I was coming in and that the hospitalist team was ready to admit me. You missed the very key point when I talkedâ?¦ it wasnt a headache, it was head pain.
The head pain was so bad, cracking my own head open by banging it on the floor seemed like it would be less painful. It was so bad that I had triple vision and could barely sign my name to paperwork. It was so bad, I passed out. It was so bad that I had a bag prepped and was willing to come back to the hospital after just leaving. But you missed all of that because you didnt listen.
You may have had a bad day or had a lot of people who could have been seen elsewhere, but saying every doctor tells their patient to come to emergency, that doesnt mean you need to wasnt helpful. If my doctor tells me to go to the ER, what am I supposed to do? You have the medical degrees.
I ended up leaving that day against medical advice (AMA). Why AMA? Because the resident said I could die by morning if I wasnt evaluated more, and you wouldnt sign off. You told me over and over I shouldnt be there but wouldnt discharge me. So I left because dying at home on my bathroom floor was a more acceptable option than dealing with you telling me how wrong I was for five more minutes. Does that make you feel like a better, doctor?
I get that I have a rare disease. I get that you dont come across it much. But why do you get to decide I am wrong when other people who would be taking care of me once Im admitted have said I need to be there?
You may have been horrible to me, but you made me a stronger patient. When I returned a few days later and was admitted for the treatment I didnt get when you were in charge of me, I was strong enough to say no when they wanted to discharge me. I knew I wasnt well enough and they hadnt found the cause, so I made them transfer me.
While you thought I was being over dramatic, which clouded my treatment, I made them transfer me to a hospital where they did find another problem a problem that still may have been hiding today if you hadnt broken me down so much that I started saying the white coat does not mean the person has common sense.
You broke me to a place where I no longer take a doctors word without finding out myself. While it may be slow progress in treatments, you taught me I cant totally trust my care to a doctor without learning myself because there will always people like you around. There will always be a doctor who makes up his or her mind before even seeing the patient. There will always that possibility that I will run into someone exactly like you, and I will no longer accept it.
You may have broken me to tears once, but you taught me I need to trust myself before I trust the white coat. (But you will still never treat me alone because I have zero trust in you.)
Thank you!
#NPAW 2017 Cookie & Cake Anyone?
Monday - April 17, 2017 @ 07:00:08
Cookie Cake anyone?
The Fleegel Family have participated in T.V. shows, FDA presentations and even decorated their very own porphyria cookie cake. YUM!!
There are all sorts of ways to enhance awareness in your community!
What are you going to do to raise porphyria awareness? We want YOU to get involved.
The APF will help you accomplish your own activity by providing:
Porphyria Brochures,
Porphyria fact sheets,
Porphyria Live DVDs,
Information to gain media attention,
PowerPoint presentations and MUCH MORE!
Contact the APF TODAY at 1.866.APF.3635 or 713.266.9617
"Remember.Research is the key to your cure!"
NPAW 2017 Merchandise Has Arrived! Place your order today!
Friday - April 14, 2017 @ 07:00:14
We are pleased to announce that the official APF T-Shirts have arrived!
Our hope for the T-shirts is that you all will buy and wear them to help promote and raise questions about Porphyria and to raise funds for the Dr. Packets, pain Management docs, among many other publications we make available at no cost.
Amy Chapman is heading up the T-shirt project. There are also some copies of Porphyria Live for sale. For information on how to order the T-Shirts and/or the Porphyria Live Video see below.
Porphyria Live DVD Introduction, Official APF T-shirts, Baseball caps & Wristbands
*The APF will not take orders or calls about T-shirts*
You can order a T-shirt and/or Porphyria Live DVD/Wristbands by sending an email order to:amy.apf@gmail.com.
Please send an email and include the following: Full Name, complete mailing address, phone number.
Order: Please send your items that you would like to order by quantity and then size for each T-shirt. For privacy purposes, please send in the email either the Visa/MC Number Expiry Date and CVV code on the back of your card. IF you prefer I can contact you by phone. NO CC info is kept on file!
Once I receive the order I will ship out your product.
All products will be shipped out Priority mail with tracking. Each person also will receive a receipt with T-shirts/DVD.
T-shirts come in the following sizes S, M, L, XL, 2 XL, 3 XL they are 100% cotton, we now have Youth sizes S, M, L. Heavy not thin fabric and very durable (EPP, XLP, CEP, HCP, VP) The cost for the New T-shirts are 20.00 each Adult or Youth
This covers the shirt, shipping and tracking, and priority mail for US orders. International orders: I will have to consult first with the post office for shipping rates.
The price of the Porphyria Live DVD is $10.00 each. Shipping is included in the price for US orders. International orders: I will have to consult first with the post office for shipping rates.
Wristbands with imprinted APF logo~ on them. Available Purple. $3.00 each or 2 for $6.00, this includes shipping & tracking. NO LATEX
Baseball caps~ APF Logo 10.00 each (adjustable strap to fit all)
Car or Fridge Magnet~ APF Logo 8.00 each
100% of all funds received will go back to the APF! Click on the link here: http://www.porphyriafoundation.com/content/official-apf-t-shirts-have-arrived
What's Your Story?
Wednesday - April 12, 2017 @ 07:00:09
What's your story?
WINK News, Southwest Florida's News Leader has chosen to highlight
Sharon Dill's story on her fight with Porphyria and the challenges that many
The porphyrias are a group of different diseases, each caused by a specific abnormality in the heme production process. Heme is a chemical compound that contains iron and gives blood its red color. The essential functions of heme depend on its ability to bind oxygen. Heme is incorporated into hemoglobin, a protein that enables red blood cells to carry oxygen from the lungs to all parts of the body. Heme also plays a role in the liver where it assists in breaking down chemicals (including some drugs and hormones) so that they are easily removed from the body. Heme is produced in the bone marrow and liver through a complex process controlled by eight different enzymes. As this production process of heme progresses, several different intermediate compounds (heme precursors) are created and modified. If one of the essential enzymes in heme production is deficient, certain precursors may accumulate in tissues (especially in the bone marrow or liver), appear in excess in the blood, and get excreted in the urine or stool. The specific precursors that accumulate depend on which enzyme is deficient. Porphyria results in a deficiency or inactivity of a specific enzyme in the heme production process, with resulting accumulation of heme precursors.
The signs and symptoms of porphyria vary among types. Some types of porphyria (called cutaneous porphyria) cause the skin to become overly sensitive to sunlight. Areas of the skin exposed to the sun develop redness, blistering and often scarring. The symptoms of other types of porphyria (called acute porphyrias) affect the nervous system. These symptoms include chest and abdominal pain, emotional and mental disorders, seizures and muscle weakness. These symptoms often appear quickly and last from days to weeks. Some porphyrias have a combination of acute symptoms and symptoms that affect the skin. Environmental factors can trigger the signs and symptoms of porphyria. These include:
Porphyria is diagnosed through blood, urine, and stool tests, especially at or near the time of symptoms. Diagnosis may be difficult because the range of symptoms is common to many disorders and interpretation of the tests may be complex. A large number of tests are available, however, but results among laboratories are not always reliable.
Each form of porphyria is treated differently. Treatment may involve treating with heme, giving medicines to relieve the symptoms, or drawing blood. People who have severe attacks may need to be hospitalized.
Most of the porphyrias are inherited conditions. The genes for all the enzymes in the heme pathway have been identified. Some forms of porphyria result from inheriting one altered gene from one parent (autosomal dominant). Other forms result from inheriting two altered genes, one from each parent (autosomal recessive). Each type of porphyria carries a different risk that individuals in an affected family will have the disease or transmit it to their children. Porphyria cutanea tarda (PCT) is a type of porphyria that is most often not inherited. Eighty percent of individuals with PCT have an acquired disease that becomes active when factors such as iron, alcohol, hepatitis C virus (HCV), HIV, estrogens (such as those used in oral contraceptives and prostate cancer treatment), and possibly smoking, combine to cause an enzyme deficiency in the liver. Hemochromatosis, an iron overload disorder, can also predispose individuals to PCT. Twenty percent of individuals with PCT have an inherited form of the disease. Many individuals with the inherited form of PCT never develop symptoms. If you or someone you know has porphyria, we recommend that you contact a genetics clinic to discuss this information with a genetics professional. To find a genetics clinic near you, contact your primary doctor for a referral.
Porphyria can be triggered by drugs (barbiturates, tranquilizers, birth control pills, sedatives), chemicals, fasting, smoking, drinking alcohol, infections, emotional and physical stress, menstrual hormones, and exposure to the sun. Attacks of porphyria can develop over hours or days and last for days or weeks.
The porphyrias have several different classification systems. The most accurate classification is by the specific enzyme deficiency. Another classification system distinguishes porphyrias that cause neurologic symptoms (acute porphyrias) from those that cause photosensitivity (cutaneous porphyrias). A third classification system is based on whether the excess precursors originate primarily in the liver (hepatic porphyrias) or primarily in the bone marrow (erythropoietic porphyrias). Some porphyrias are classified as more than one of these categories.
The cutaneous porphyrias affect the skin. People with cutaneous porphyria develop blisters, itching, and swelling of their skin when it is exposed to sunlight. The cutaneous porphyrias include the following types: Also called congenital porphyria. This is a rare disorder that mainly affects the skin. It results from low levels of the enzyme responsible for the fourth step in heme production. It is inherited in an autosomal recessive pattern. An uncommon disorder that mainly affects the skin. It results from reduced levels of the enzyme responsible for the eighth and final step in heme production. The inheritance of this condition is not fully understood. Most cases are probably inherited in an autosomal dominant pattern, however, it shows autosomal recessive inheritance in a small number of families. A rare disorder that mainly affects the skin. It results from very low levels of the enzyme responsible for the fifth step in heme production. It is inherited in an autosomal recessive pattern. A rare disorder that can have symptoms of acute porphyria and symptoms that affect the skin. It results from low levels of the enzyme responsible for the sixth step in heme production. It is inherited in an autosomal dominant pattern. The most common type of porphyria. It occurs in an estimated 1 in 25,000 people, including both inherited and sporadic (noninherited) cases. An estimated 80 percent of porphyria cutanea tarda cases are sporadic. It results from low levels of the enzyme responsible for the fifth step in heme production. When this condition is inherited, it occurs in an autosomal dominant pattern. A disorder that can have symptoms of acute porphyria and symptoms that affect the skin. It results from low levels of the enzyme responsible for the seventh step in heme production. It is inherited in an autosomal dominant pattern.
The acute porphyrias affect the nervous system. Symptoms of acute porphyria include pain in the chest, abdomen, limbs, or back; muscle numbness, tingling, paralysis, or cramping; vomiting; constipation; and personality changes or mental disorders. These symptoms appear intermittently. The acute porphyrias include the following types: This is probably the most common porphyria with acute (severe but usually not long-lasting) symptoms. It results from low levels of the enzyme responsible for the third step in heme production. It is inherited in an autosomal dominant pattern. A very rare disorder that results from low levels of the enzyme responsible for the second step in heme production. It is inherited in an autosomal recessive pattern.
National Porphyria Awareness Week: April 22 - 29, 2017
The challenge of living with porphyria starts with how little is known about it among friends, family and the medical community. That is why National Porphyria Awareness Weekis so important. It provides each of YOU with the opportunity to enhance porphyria awareness in your sphere of influence. Here are some suggestions for ways that you can help:
*SHARE knowledge about porphyria at your doctor's offices and local hospitals. You might suggest that they host a seminar or grand rounds on porphyria; ask if there is a local meeting where you can hand out materials.
*ASSIST at medical conventions or health fairs to educate family, friends, and physicians on porphyria.
*Tell your story to local media. Television, newspapers and community magazines are looking for human interest stories about people who have encountered a major illness and have undertaken the challenge.
*VOLUNTEER your talents or skills to help achieve the educational programs of the APF. For example, donate one of your paintings, sculpture, computer expertise, business acumen etc. for our fall raffle or to help APF.
*HOLD a community race, car wash or other fund raising activity.
*HONOR your loved one with a gift to the APF for a birthday, anniversary, holiday or memorial tribute.
*WRITE a letter to friends and family asking them to consider making a donation;
The APF can help you accomplish this goal by providing materials:
What are you going to do to raise awareness in your community? We want YOU to get involved.
The APF will help you accomplish your own activity by providing:
Porphyria Brochures,
Porphyria fact sheets,
Porphyria Live DVDs,
Information to gain media attention,
PowerPoint presentations and MUCH MORE!
Call the APF TODAY! 1.866.APF.3635
"Remember.Research is the key to your cure!"
Effective Management of Acute Intermittent Porphyria: Part 3
Wednesday - March 29, 2017 @ 09:00:27
Effective Management of Acute Intermittent Porphyria: Patients Deserve Access to Appropriate Care
What is Acute Intermittent Porphyria (AIP)? AIP is one of a group of rare, inherited disorders called porphyrias that all involve the overproduction and buildup of chemicals called porphyrins or porphyrin precursors. AIP is
Patients Diagnosed with AIP Deserve Access to Treatment For people living with Acute Intermittent Porphyria (AIP), there are treatments available, but many still encounter barriers to receiving accessible care at their preferred hospitals.
The American Porphyria Foundation (APF) urges all healthcare institutions to provide timely access to medically necessary care. We encourage everyone involved in the care of patients with AIP to demand timely access to FDA-approved treatments. We believe:
Regulators should advocate for patient access to approved treatments Providers need to understand AIP and provide prompt, appropriate treatment Patients deserve access and coverage for available, life-saving treatment options deaminase) and is characterized by acute attacks, often triggered by environmental factors or hormone changes. In some cases, the cause
Because the symptoms of AIP mimic other common conditions, the diagnostic process can be long suffering for years before receiving a proper diagnosis. â?¢ Severe, intense abdominal pain â?¢ Nausea and vomiting â?¢ Constipation â?¢ Diarrhea â?¢ Pain in the extremities, back, chest, neck,or head â?¢ Muscle weakness â?¢ Convulsions â?¢ Mental symptoms â?¢ Respiratory paralysis â?¢ Fast heart beat â?¢ High blood pressure â?¢Dark reddish or purple urine
This educational material is provided by the American Porphyria Foundation. For more information,visit porphyriafoundation.com
Managing AIP AIP is a genetic disease and there is no cure. However, there are strategies that can be used to prevent the onset or severity of attacks:
â?¢ Avoiding dieting or fasting, even for short periods â?¢ Avoiding certain medications that can trigger an attack â?¢ Limiting physical and emotional stress â?¢ Avoiding alcohol
People with frequent attacks can also consider wearing a medical alert bracelet.
References 1. Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142:439-450.
2. Bonkovsky HL, et al. Acute porphyrias in the USA: features of 108 subjects from porphyrias consortium. The Amer. Jrnl of Med. 2014;127: 1233-1241.
3. Crimlisk HL. The little imitator-porphyria: a neuropsychiatric disorder. J Neurol Neurosurg Psychiatry. 1997;62:319-328.
Prompt Treatment is Critical Even with proper diet and control of environmental factors, some patients still have recurring, and often severe attacks. Acute attacks typically require hospitalization and delays in treatment can be life-threatening and result in serious complications such as irreversible nerve damage.
Treatments can include: â?¢ Medication to manage pain and other symptoms â?¢ Glucose and carbohydrates given orally or intravenously â?¢ Intravenous heme if the patients symptoms fail to improve within 36 hours of glucose treatment
What's all in the New Acute Porphyria Tool Kit? (AHP) Part 2
Monday - March 27, 2017 @ 09:00:18
What's all in the New Acute Porphyria Tool Kit? (AHP) How can it benefit me or a caregiver? How will it affect my Doctor's appointments? All these tools come free of charge and can be downloaded any time. Please click on the link here: Access to Care Toolkit:
Access to Care Toolkit for the Acute Porphyrias is now available
A downloadable Access to Care Toolkit is a resource designed to help patients living with Acute Intermittent Porphyria (AIP), Hereditary Coproporphyria (HCP), and Variegate Porphyria (VP) or their caregivers, loved ones and healthcare providers secure access to Panhematin at their preferred health facility. We have recently learned of patients who are being denied this treatment from some hospitals and directed to secure another healthcare provider. If this has happened to you or someone you know or care for, please use these tools to request help from your state and local representatives and health advocacy organizations. We understand the debilitating effects of acute porphyria and hope these resources will help you secure access to Panhematin when you need it most.
The Toolkit contains the following materials:
*Healthcare Conversation Tracker is a simple form to record your conversations with doctors, insurance agents, etc.
*Customizable letter templates to record your details to use for doctors, state departments, insurance, etc.
*Access to Care Fact Sheet defines AIP, its symptoms and why it's important for patients to get immediate care
*Patient Bill of Rights can be used to support your appeal for access to treatment
This Toolkit can be found on the APF website. Contact the APF office today if you have questions!
Part 2~
Effective Management of Acute Intermittent Porphyria: Healthcare Conversation Tracker Instructions It is helpful to keep detailed records of your conversations and interactions with doctors, nurses, counselors, and any other healthcare staff. It is also important to keep track of your conversations with insurance providers. This information can support your appeals for access to care and treatment and should be provided with your letter. Be sure to take notes on all your healthcare visits including the date, time, and outcomes, including any incidents or access to care issues. Also note any phone conversations. _____________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________ Be as much of the conversation, if possible. This will make it easier to remember. ____________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________ Also note conversations with your insurance provider and include the name of the representative. If you have to leave a telephone message, it is helpful to record the date, time, and persons name.__________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
This educational material is provided by the American Porphyria Foundation. For more information, visit porphyriafoundation.com Use the form below to detail your issues with obtaining access to care and treatment for your condition, or reimbursement. Print multiple copies and record each visit or phone call you make. If you are unable to take notes, ask a relative, friend or caregiver if they can help. ___________________________________________________________________________________________________________________________________________________________________________________________________________________________ Submit a copy of this document along with other documents to support your claims. Remember to be thorough and capture as many details as possible. Health Visit/Phone Call Report:_______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________ Date:____________________________________________________________________ Time:____________________________________________________________________ Address: ____________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________ I went in or called because:_____________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________ I spoke to a (check one): Doctor Nurse Counselor Administrative Staff His/Her name was:______________________________________________________________________________________________________________________________________________ I also interacted with a (check one): Doctor Nurse Counselor Administrative staff I spoke with my insurance provider: __________________________________________________________________________________________________________________________________________________ The representative's name was: He/She told me:____________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________ Other Notes:_______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Access to Care Toolkit for the Acute Porphyrias is now available Part 1
Friday - March 24, 2017 @ 09:00:14
What's all in the New Acute Porphyria Tool Kit? (AHP) How can it benefit me or a caregiver? How will it affect my Doctor's appointments? All these tools come free of charge and can be downloaded any time. Please click on the link here: Access to Care Toolkit:
Access to Care Toolkit for the Acute Porphyrias is now available
A downloadable Access to Care Toolkit is a resource designed to help patients living with Acute Intermittent Porphyria (AIP), Hereditary Coproporphyria (HCP), and Variegate Porphyria (VP) or their caregivers, loved ones and healthcare providers secure access to Panhematin at their preferred health facility. We have recently learned of patients who are being denied this treatment from some hospitals and directed to secure another healthcare provider. If this has happened to you or someone you know or care for, please use these tools to request help from your state and local representatives and health advocacy organizations. We understand the debilitating effects of acute porphyria and hope these resources will help you secure access to Panhematin when you need it most.
The Toolkit contains the following materials:
*Healthcare Conversation Tracker is a simple form to record your conversations with doctors, insurance agents, etc.
*Customizable letter templates to record your details to use for doctors, state departments, insurance, etc.
*Access to Care Fact Sheet defines AIP, its symptoms and why it's important for patients to get immediate care
*Patient Bill of Rights can be used to support your appeal for access to treatment
This Toolkit can be found on the APF website. Contact the APF office today if you have questions!
Part 1~
Sample Letter from Patient
Note: The letter provided is only a sample providing suggestions to the writer for composing his/her own letter. It is the writers responsibility to detail his/her own thoughts and experiences in a personally acceptable manner. All content is ultimately the responsibility of the writer.
Dear [insert name],
I am a resident of [insert city or town] who lives with a serious medical condition known as Acute Intermittent Porphyria (AIP). AIP is a rare disease that is debilitating, painful and has profoundly impacted my life. I struggled for years to obtain a diagnosis. However, with my diagnosis, I was finally able to manage my disease with Panhematin, which I received at [insert name of hospital.]
I was devastated to learn that [insert name of hospital] will no longer honor my physicians prescription and provide me with access to this treatment. Instead, I was told to find it through another hospital.
As a patient living with a rare disease, it is important I continue to receive timely access to the most effective treatment available. During an attack, I experience excruciating pain and [insert specific symptoms], that make it extremely difficult to find another hospital, much less get myself there.
Living with AIP is extremely challenging and recurrent, severe attacks have limited my ability to work and provide for myself and my family. It is extremely important that I have access to Panhematin at my preferred hospital. I worry that delays in treatment will result in a serious attack leading to hospitalization, and serious, potentially life-threatening complications.
I appeal to you to help me in urging [name of hospital] to provide continued access to the treatment I desperately need.
This is in keeping with my rights as a patient and in accordance with [insert name of state] Patients Bill of Rights and Responsibilities, which state:
[insert exact language from bill of rights]
[insert exact language from bill of rights]
I appreciate your thoughtful consideration of my appeal and look forward to hearing from you soon.
Sincerely,
[Insert your name]
Sample Letter from Caregiver
Note: The letter provided is only a sample providing suggestions to the writer for composing his/her own letter. It is the writers responsibility to detail his/her own thoughts and experiences in a personally acceptable manner. All content is ultimately the responsibility of the writer.
Dear [name],
Imagine you, or a loved one, suffered from a potentially life-threatening rare disease only to have the hospital you know and trust deny access to the only treatment that helps.
Unfortunately, this is not a hypothetical story. This is my reality. I am a resident of [insert city or town] and I provide care to a person living with Acute Intermittent Porphyria (AIP), a rare, often debilitating and painful disease, that can lead to hospitalization if untreated. He/she] has been receiving Panhematin, the only FDA approved hospital-based treatment for AIP for [insert number] years.
We were recently devastated to learn that [name of hospital] will no longer provide access to Panhematin.In fact, the [insert title of person you spoke with] turned us away and told us to find another hospital for treatment.
This is extremely troubling and disappointing, because it is critical that AIP patients have prompt, access to treatment. Delays can result in a severe, potentially life-threatening attack. Left untreated, an attack can result in serious complications such as irreversible nerve damage.
As a caregiver, the thought of watching my loved one suffer needlessly when there is a treatment available at their preferred hospital is unacceptable. I appeal to [insert organization/person receiving letter] to reconsider this misguided decision and continue to give patients the treatment they need and deserve in accordance with [insert name of state] Patients Bill of Rights and Responsibilities, which state:
[insert exact language from bill of rights]
[insert exact language from bill of rights]
I appreciate your thoughtful consideration and look forward to hearing from you soon.
Sincerely,
[Insert your name]
Sample Letter from Healthcare Provider
Note: The letter provided is only a sample providing suggestions to the writer for composing his/her own letter. It is the writers responsibility to detail his/her own thoughts and experiences in a personally acceptable manner. All content is ultimately the responsibility of the writer.
Dear [insert name],
As the treating physician for a patient living with Acute Intermittent Porphyria (AIP), a rare disorder that often leads to extreme pain and debilitating symptoms, I was disappointed to learn that [name of hospital] will no longer provide [him/her] access to Panhematin, the only FDA-approved, hospital-based treatment for AIP attacks.
This is extremely troubling and I appeal to [insert organization/person receiving letter] to help ensure [name of hospital] provides continued access to this critical treatment in the interest of my patients health, and in keeping with [his/her] rights as a patient. Patients with AIP require prompt, unrestricted access to Panhematin that is prescribed under the care of physicians experienced in the management of porphyria, at hospitals that offer the necessary clinical and laboratory diagnostic and monitoring techniques. Delays in treatment can result in severe, potentially life-threatening attacks. Left untreated, attacks can lead to hospitalization, irreversible nerve damage and possibly death.
As healthcare providers to the community, [name of hospital] has an obligation to provide timely treatment to patients. Patients with AIP often experience excruciating pain, which greatly reduces their ability to manage changes in treatment schedules and locations. Turning my patient away from this facility is more than an inconvenience and could cause long-term health issues.
I appreciate your thoughtful consideration of my appeal and look forward to your reply.
Sincerely,
[Insert name]
[Insert title]
National Porphyria Awareness Week (NPAW) is right around the corner.
Wednesday - March 22, 2017 @ 14:26:52
National Porphyria Awareness Week (NPAW) is right around the corner.
NPAW: April 22 - 29, 2017
See the story below as a great example of how to raise awareness in your community.
For the second year in a row, the Cook family put on a fantastic benefit barrel race. As you may know, the Cook brothers, Cason and Caul, have EPP and have set a great example about enhancing awareness of the disease in their local area. They have been hosting a Hat Day, supporting the APF and porphyria awareness, for many years in their hometown of Vernon, Texas.
There were over 159 entries for the race and an incredible outpouring of support from the community with volunteers and sponsors.
Watch for stories like this from individuals and families who have changed the world of Porphyria Awareness in the community.
It is our hope that you will become involved in this year's awareness
activities.
Let's RAISE PORPHYRIA AWARENESS!
"Remember.Research is the key to your cure!"
Access to Care Toolkit for the Acute Porphyrias is now available
Wednesday - March 15, 2017 @ 09:30:08
Access to Care Toolkit for the Acute Porphyrias is now available
A downloadable Access to Care Toolkit is a resource designed to help patients living with Acute Intermittent Porphyria (AIP), Hereditary Coproporphyria (HCP), and Variegate Porphyria (VP) or their caregivers, loved ones and healthcare providers secure access to Panhematin at their preferred health facility. We have recently learned of patients who are being denied this treatment from some hospitals and directed to secure another healthcare provider. If this has happened to you or someone you know or care for, please use these tools to request help from your state and local representatives and health advocacy organizations. We understand the debilitating effects of acute porphyria and hope these resources will help you secure access to Panhematin when you need it most.
The Toolkit contains the following materials:
*Healthcare Conversation Tracker is a simple form to record your conversations with doctors, insurance agents, etc.
*Customizable letter templates to record your details to use for doctors, state departments, insurance, etc.
*Access to Care Fact Sheet defines AIP, its symptoms and why it's important for patients to get immediate care
*Patient Bill of Rights can be used to support your appeal for access to treatment
This Toolkit can be found on the APF website. Contact the APF office today if you have questions!
Alnylam Receives European Medicines Agency PRIME Designation for Accelerated Assessment of Givosiran, an Investigational RNAi Therapeutic for the Treatment of Acute Hepatic Porphyrias
Monday - March 13, 2017 @ 09:30:15
Alnylam Receives European Medicines Agency PRIME Designation for Accelerated Assessment of Givosiran, an Investigational RNAi Therapeutic for the Treatment of Acute Hepatic Porphyrias
03.01.2017
CAMBRIDGE, Mass.--(BUSINESS WIRE)-- Alnylam Pharmaceuticals, Inc. (Nasdaq:ALNY), the leading RNAi therapeutics company, announced today that the European Medicines Agency (EMA) has granted access to its Priority Medicines (PRIME) scheme for givosiran (ALN-AS1), an investigational RNAi therapeutic targeting aminolevulinic acid synthase 1 (ALAS1) for the treatment of acute hepatic porphyrias. The purpose of the PRIME initiative is to bring treatments to patients faster by enhancing the EMA's support for the development of medicines for diseases where there is an unmet medical need and where early clinical data show potential to benefit patients.
Promising results from the Phase 1 study of givosiran formed the basis of the application for PRIME. The ongoing Phase 1 trial is being conducted as a randomized, double-blind, placebo-controlled study. Specifically, data were recently reported in patients with acute intermittent porphyria (AIP) experiencing recurrent attacks. As presented at the 2016 American Society of Hematology (ASH) meeting, givosiran demonstrated initial evidence for clinical activity in AIP patients with meaningful reductions in the number and frequency of porphyria attacks. In the first two dose cohorts, givosiran was found to be generally well tolerated with no drug-related serious adverse events. In the third dose cohort, which remains blinded, one death due to acute pancreatitis, considered unlikely related to givosiran or placebo, was reported after the data transfer date.
"We are pleased to have givosiran accepted into the PRIME program. We believe givosiran could be a potentially transformative treatment option for patients with acute hepatic porphyrias, a family of debilitating and life threatening diseases with enormous unmet medical need," said Jeff Miller, Vice President, General Manager, Givosiran Program at Alnylam. "We look forward to collaborating with the EMA on the accelerated assessment of givosiran, with the goal of advancing this investigational medicine into a Phase 3 trial in late 2017."
Givosiran has previously been granted Orphan Drug Designations in both the EU and the U.S. for the treatment of acute hepatic porphyrias. Through the PRIME program Alnylam will have enhanced scientific and regulatory support from the EMA, including its advice on optimization of the development pathway and the potential for accelerated assessment of the Marketing Authorisation Application (MAA).
About Givosiran Alnylam is developing givosiran (formerly known as ALN-AS1), a subcutaneously administered, investigational RNAi therapeutic targeting aminolevulinic acid synthase 1 (ALAS1) for the treatment of acute hepatic porphyrias, including acute intermittent porphyria (AIP). AIP is an ultra-rare autosomal dominant disease caused by loss of function mutations in porphobilinogen deaminase (PBGD), an enzyme in the heme biosynthesis pathway that can result in accumulation of toxic heme intermediates, including aminolevulinic acid (ALA) and porphobilinogen (PBG). Patients with AIP can suffer from acute and/or recurrent life-threatening attacks characterized by severe abdominal pain, neuropathy (affecting the central, peripheral or autonomic nervous system), and neuropsychiatric manifestations. Givosiran is an ESC-GalNAc-siRNA conjugate targeting ALAS1, a liver-expressed, rate-limiting enzyme upstream of PBGD in the heme biosynthesis pathway. Inhibition of ALAS1 is known to reduce the accumulation of heme intermediates that cause the clinical manifestations of AIP. Givosiran has the potential to be a novel treatment approach for the prevention of recurrent attacks. Givosiran is an investigational compound, currently in early stage clinical development. The safety and efficacy of givosiran have not been evaluated by the U.S. Food and Drug Administration or any other health authority.
About Acute Hepatic Porphyrias The porphyrias are a family of rare metabolic disorders with mostly autosomal dominant inheritance predominantly caused by a genetic mutation in one of the eight enzymes responsible for heme biosynthesis. Acute hepatic porphyrias (AHP) constitute a subset where the enzyme deficiency occurs within the liver, and includes acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP). Exposure of AHP patients to certain drugs, dieting, or hormonal changes can trigger strong induction of aminolevulinic acid synthase 1 (ALAS1), another enzyme in the heme biosynthesis pathway, which can lead to accumulation of neurotoxic heme intermediates that precipitate disease symptoms. Patients with AHP can suffer from a range of symptoms that, depending on the specific type, can include acute and/or recurrent life-threatening attacks with severe abdominal pain, peripheral and autonomic neuropathy, neuropsychiatric manifestations, cutaneous lesions and possibly paralysis and death if untreated or if there are delays in treatment. There are no approved treatments for the prevention of attacks; the only approved treatment for acute attacks is hemin for injection (Panhematin or Normosang), a preparation of heme derived from human blood. Hemin requires administration through a large vein or a central intravenous line and is associated with a number of complications including thrombophlebitis or coagulation abnormalities. Chronic administration of hemin may result in renal insufficiency, iron overload, systemic infections (due to the requirement for central venous access) and, in some instances, tachyphylaxis.
About GalNAc Conjugates and Enhanced Stabilization Chemistry (ESC)-GalNAc Conjugates GalNAc-siRNA conjugates are a proprietary Alnylam delivery platform and are designed to achieve targeted delivery of RNAi therapeutics to hepatocytes through uptake by the asialoglycoprotein receptor. Alnylam's Enhanced Stabilization Chemistry (ESC)-GalNAc-conjugate technology enables subcutaneous dosing with increased potency and durability, and a wide therapeutic index. This delivery platform is being employed in nearly all of Alnylam's pipeline programs, including programs in clinical development.
About RNAi RNAi (RNA interference) is a revolution in biology, representing a breakthrough in understanding how genes are turned on and off in cells, and a completely new approach to drug discovery and development. Its discovery has been heralded as "a major scientific breakthrough that happens once every decade or so," and represents one of the most promising and rapidly advancing frontiers in biology and drug discovery today which was awarded the 2006 Nobel Prize for Physiology or Medicine. RNAi is a natural process of gene silencing that occurs in organisms ranging from plants to mammals. By harnessing the natural biological process of RNAi occurring in our cells, the creation of a major new class of medicines, known as RNAi therapeutics, is on the horizon. Small interfering RNA (siRNA), the molecules that mediate RNAi and comprise Alnylam's RNAi therapeutic platform, target the cause of diseases by potently silencing specific mRNAs, thereby preventing disease-causing proteins from being made. RNAi therapeutics have the potential to treat disease and help patients in a fundamentally new way.
About Alnylam Pharmaceuticals Alnylam is a biopharmaceutical company developing novel therapeutics based on RNA interference, or RNAi. The company is leading the translation of RNAi as a new class of innovative medicines. Alnylam's pipeline of investigational RNAi therapeutics is focused in 3 Strategic Therapeutic Areas (STArs): Genetic Medicines, with a broad pipeline of RNAi therapeutics for the treatment of rare diseases; Cardio-Metabolic Disease, with a pipeline of RNAi therapeutics toward genetically validated, liver-expressed disease targets for unmet needs in cardiovascular and metabolic diseases; and Hepatic Infectious Disease, with a pipeline of RNAi therapeutics that address the major global health challenges of hepatic infectious diseases. In early 2015, Alnylam launched its "Alnylam 2020" guidance for the advancement and commercialization of RNAi therapeutics as a whole new class of innovative medicines. Specifically, by the end of 2020, Alnylam expects to achieve a company profile with 3 marketed products, 10 RNAi therapeutic clinical programs - including 4 in late stages of development - across its 3 STArs. The company's demonstrated commitment to RNAi therapeutics has enabled it to form major alliances with leading companies including Ionis, Novartis, Roche, Takeda, Merck, Monsanto, The Medicines Company, and Sanofi Genzyme. In addition, Alnylam holds an equity position in Regulus Therapeutics Inc., a company focused on discovery, development, and commercialization of microRNA therapeutics. Alnylam scientists and collaborators have published their research on RNAi therapeutics in over 200 peer-reviewed papers, including many in the world's top scientific journals such as Nature, Nature Medicine, Nature Biotechnology, Cell, New England Journal of Medicine, and The Lancet. Founded in 2002, Alnylam maintains headquarters in Cambridge, Massachusetts. For more information about Alnylam's pipeline of investigational RNAi therapeutics, please visit www.alnylam.com.
Alnylam Forward Looking Statements Various statements in this release concerning Alnylam's future expectations, plans and prospects, including without limitation, Alnylam's views with respect to the potential for RNAi therapeutics, including givosiran, its expectations regarding the timing of clinical studies, including the initiation of a Phase 3 trial for givosiran following interactions with regulatory authorities, its expectations regarding scientific and regulatory support for givosiran from the EMA and collaborating with the EMA on the accelerated assessment of givosiran, its expectations regarding its STAr pipeline growth strategy, and its "Alnylam 2020" guidance for the advancement and commercialization of RNAi therapeutics, constitute forward-looking statements for the purposes of the safe harbor provisions under The Private Securities Litigation Reform Act of 1995. Actual results and future plans may differ materially from those indicated by these forward-looking statements as a result of various important risks, uncertainties and other factors, including, without limitation, Alnylam's ability to discover and develop novel drug candidates and delivery approaches, successfully demonstrate the efficacy and safety of its product candidates, the pre-clinical and clinical results for its product candidates, which may not be replicated or continue to occur in other subjects or in additional studies or otherwise support further development of product candidates for a specified indication or at all, actions or advice of regulatory agencies, which may affect the design, initiation, timing, continuation and/or progress of clinical trials or result in the need for additional pre-clinical and/or clinical testing, delays, interruptions or failures in the manufacture and supply of our product candidates, obtaining, maintaining and protecting intellectual property, Alnylam's ability to enforce its intellectual property rights against third parties and defend its patent portfolio against challenges from third parties, obtaining and maintaining regulatory approval, pricing and reimbursement for products, progress in establishing a commercial and ex-United States infrastructure, competition from others using technology similar to Alnylam's and others developing products for similar uses, Alnylam's ability to manage its growth and operating expenses, obtain additional funding to support its business activities, and establish and maintain strategic business alliances and new business initiatives, Alnylam's dependence on third parties for development, manufacture and distribution of products, the outcome of litigation, the risk of government investigations, and unexpected expenditures, as well as those risks more fully discussed in the "Risk Factors" filed with Alnylam's most recent Annual Report on Form 10-K filed with the Securities and Exchange Commission (SEC) and in other filings that Alnylam makes with the SEC. In addition, any forward-looking statements represent Alnylam's views only as of today and should not be relied upon as representing its views as of any subsequent date. Alnylam explicitly disclaims any obligation, except to the extent required by law, to update any forward-looking statements.
The scientific information referenced in this news release relating to givosiran is preliminary and investigative. Givosiran has not been approved by the U.S. Food and Drug Administration, European Medicines Agency, or any other regulatory authority and no conclusions can or should be drawn regarding its safety or effectiveness.
Why Join a Clinical Study?
Friday - March 10, 2017 @ 09:30:16
Clinical Studies
There are two main types of clinical studies: clinical trials and observational studies. In a clinical trial, there is some form of treatment intervention. There is no intervention in an observational study, which is aimed at observing patients to better understand the long-term course of their disease.
Clinical trials are used to test new treatments before they are approved for use by the FDA. This type of trial gives patients a chance to try out a new medication in its early stages. As with any experiment, the result of a trial is not known before its conclusion. Your participation could help demonstrate a terrific treatment breakthrough, or it could help scientists discover that a new treatment does not work after all. There may be some risk involved from the treatment in a clinical trial.
Participating in either a clinical trial or an observational study is a serious responsibility. Volunteering to participate could be a way to help yourself, affected family members and other patients by advancing medical and scientific knowledge of your condition. Some patients derive great satisfaction from assisting doctors in the study of their disease. Participation in a study can also mean a chance to meet a porphyria researcher in a clinical setting, and the consultation can be beneficial.
My name is Kelly Story and I live with my husband, Chad, and two cats in Kissimmee, FL. In July of 1999, one month after our wedding, I was out of town on business, and I noticed tiny little water blisters all over my hands. The blisters didn't itch and were not painful. Although I found them to be very odd, I didn't worry much.
Over a short period of time, my skin became extremely fragile. It seemed like almost anything would cause a scrape on the skin of my hands. Plus, the blisters got much worse. My hands looked like something out of a horror movie. I was so embarrassed all of the time, and I cried a lot. I went to a dermatologist. After two visits, he told me that he was fairly certain that I had PCT but suggested that I go to a specialist. After several humbling tests and weeks of waiting, I was officially diagnosed with PCT.
The doctor said that I would have to start phlebotomies. This meant that over a pint of blood would be taken from me at one time. Even after I began the phlebotomy treatments, the blisters moved to my arms. These blisters itched tremendously. My poor husband didn't know what was happening, because, I would wake up in the middle of the night with a frenzy of scratching and crying. The itching was unbearable. I would feel so guilty afterwards. After all, I was receiving treatment for my condition and knew that I did not have a fatal disease. There are other people in the world with so much more serious problems. But, it was still uncomfortable, and my hands and arms were just so ugly.
I was tired a lot during the six month or so period I was having the phlebotomies. The doctor said that I would most likely be temporarily anemic. This was the result of having the blood taken from me. I used to do aerobic exercises at least four times a week, but that stopped. I also got headaches quite often. Finally, I was in remission. No more cuts, no more blisters, and no more itching.
I lived for a while with the dark scars on my hands from all of the sores and blisters and several lighter ones on my arms. But now, the scars have faded tremendously. You can't see any on my arms, and the ones on my hands are very pale. It may sound vain, but I never thought I would have pretty hands again, and now I do. To this day, I still cringe if I accidentally knock my hand into something. But, I am always relieved when I look down and see no scrape or cut.
Since being diagnosed with PCT, I've taken estrogen and alcohol out of my life, and I try my best to stay out of the sun. It's hard living in Florida, but I just keep applying that good old sunscreen. I'm very blessed to have such a supportive husband. I told Chad on our first date that I was high maintenance. Boy, neither one of us knew how much!
Porphyria cutanea tarda (PCT) is the most common subtype of porphyria.[1] The disease is named because it is a porphyria that often presents with skin manifestations later in life. The disorder results from low levels of the enzyme responsible for the fifth step in heme production. Heme is a vital molecule for all of the body's organs. It is a component of hemoglobin, the molecule that carries oxygen in the blood.
Hepatoerythropoietic porphyria has been described as a homozygous form of porphyria cutanea tarda,[2] although it can also be caused if two different mutations occur at the same locus.
Porphyria cutanea tarda (commonly referred to as PCT) is recognized as the most prevalent subtype of porphyritic diseases.[3]
The disease is characterized by onycholysis and blistering of the skin in areas that receive higher levels of exposure to sunlight. The primary cause of this disorder is a deficiency of uroporphyrinogen decarboxylase (UROD), a cytosolic enzyme that is a step in the enzymatic pathway that leads to the synthesis of heme. While a deficiency in this enzyme is the direct cause leading to this disorder, there are a number of both genetic and environmental risk factors that are associated with PCT.[4]
Typically, patients who are ultimately diagnosed with PCT first seek treatment following the development of photosensitivities in the form of blisters and erosions on commonly exposed areas of the skin. This is usually observed in the face, hands, forearms, and lower legs. It heals slowly and with scarring. Though blisters are the most common skin manifestations of PCT, other skin manifestations like hyperpigmentation (as if they are getting a tan) and hypertrichosis (mainly on top of the cheeks) also occur. PCT is a chronic condition, with external symptoms often subsiding and recurring as a result of a number of factors. In addition to the symptomatic manifestation of the disease in the skin, chronic liver problems are extremely common in patients with the sporadic form of PCT. These include hepatic fibrosis (scarring of the liver), cirrhosis, and inflammation. However, liver problems are less common in patients with the inherited form of the disease.[5] Additionally, patients will often void a wine-red color urine with an increased concentration of uroporphyrin I due to their enzymatic deficiency.[6]
20% of cases of porphyria cutanea tarda are inherited in an autosomal dominant pattern.
Inherited mutations in the URODgene cause about 20% of cases (the other 80% of cases do not have mutations in UROD, and are classified as sporadic). UROD makes an enzyme called uroporphyrinogen III decarboxylase, which is critical to the chemical process that leads to heme production. The activity of this enzyme is usually reduced by 50% in all tissues in people with the inherited form of the condition.
Nongenetic factors such as alcohol abuse, excess iron, and others listed above can increase the demand for heme and the enzymes required to make heme. The combination of this increased demand and reduced activity of uroporphyrinogen decarboxylase disrupts heme production and allows byproducts of the process to accumulate in the body, triggering the signs and symptoms of porphyria cutanea tarda.
The HFE gene makes a protein that helps cells regulate the absorption of iron from the digestive tract and into the cells of the body. Certain mutations in the HFE gene cause hemochromatosis (an iron overload disorder). People who have these mutations are also at an increased risk of developing porphyria cutanea tarda.
In the 20% of cases where porphyria cutanea tarda is inherited, it is inherited in an autosomal dominant pattern, which means one copy of the altered gene is sufficient to decrease enzyme activity and cause the signs and symptoms of the disorder.
While inherited deficiencies in uroporphyrinogen decarboxylase often lead to the development of PCT, there are a number of risk factors that can both cause and exacerbate the symptoms of this disease. One of the most common risk factors observed is infection with the Hepatitis C virus.[7] One review of a collection of PCT studies noted Hepatitis C infection in 50% of documented cases of PCT. Additional risk factors include alcohol abuse, excess iron (from iron supplements as well as cooking on cast iron skillets), and exposure to chlorinated cyclic hydrocarbons and Agent Orange.
Porphyria cutanea tarda is primarily caused by uroporphyrinogen decarboxylase deficiency (UROD). Uroporphyrinogen decarboxylase occurs in nature as a homodimer of two subunits. It participates in the fifth step in heme synthesis pathway, and is active in the cytosol. This enzymatic conversion results in coproporphyrinogen III as the primary product. This is accomplished by the clockwise removal of the four carboxyl groups present in the cyclic uroporphyrinogen III molecule. Therefore, a deficiency in this enzyme causes the aforementioned buildup of uroporphyrinogen and hepta-carboxylic porphyrinogen, and to a lesser extent hexa-carboxylic porphyrinogen, and penta-carboxylic porphyrinogen in the urine, which can be helpful in the diagnosis of this disorder.[11][12]
The dermatological symptoms of PCT that include blistering and lesions on sun-exposed areas of the skin are caused by a buildup of porphyrin compounds (specifically uroporphyrinogen) close to the surface of the skin that have been oxidized by free radicals or sunlight.[13] The oxidized porphyrins initiate degranulation of dermal mast cells,[14] which release proteases that catabolize the surrounding proteins.[15] This begins a cell mediated positive feedback loop which matches the description of a type 4 delayed hypersensitivity reaction.[citation needed] The resulting blisters therefore do not appear immediately, but begin to show up 23 days after sun exposure. Due to the highly conjugated structure of porphyrins involving alternating single and double carbon bonds, these compounds exhibit a deep purple color, resulting in the discoloration observed in the skin. Excess alcohol intake decreases hepcidin production which leads to increased iron absorption from the gut and an increase in oxidative stress. This oxidative stress then leads to inhibition of uroporphyrinogen decarboxylase, creating an excess of uroporphyrinogen III which is oxidized from the relatively harmless porphyrinogens into their reduced porphyrins form.[16] Concentrated instances of oxidative stress (alcohol, physical trauma, psychological stress, etc) cause the liver to hemorrhage these porphyrins into the blood stream where they are then susceptible to oxidation. The strong association of PCT with Hepatitis C infection is not entirely understood. Studies have suggested that the cytopathic effect of the virus on hepatocytes can lead to the release of free iron. This iron can disrupt the activity of cytochrome p450, releasing activated oxygen species. These can oxidize the UROD substrate uroporphyrinogen, which can result in the inhibition of UROD and lead to deficient activity of this key enzyme.
Excess alcohol abuse is frequently associated with both inducing PCT[17] and aggravating a preexisting diagnosis of the disorder. It is thought to do so by causing oxidative damage to liver cells, resulting in oxidized species of uroporphyrinogen that inhibit the activity of hepatic UROD. It is also felt to increase the uptake of iron in liver cells, leading to further oxidation of uroporphyrinogen by the release of activated oxygen species. Additionally, exposure to chlorinated cyclic hydrocarbons can lead to a deficiency in the activity of uroporphyrinogen decarboxylase, causing the buildup of excess uroporphyrinogen. Additionally, alcohol has been shown to increase the activity of the delta-aminolevulinic acid synthetase (ALA synthetase), the rate-limiting enzymatic step in heme synthesis in the mitochondria, in rats.[18] Therefore, alcohol consumption may increase the production of uroporphyrinogen, exacerbating symptoms in individuals with porphyria cutanea tarda.
While the most common symptom of PCT is the appearance of skin lesions and blistering, their appearance does not single-handedly lead to a conclusive diagnosis. Laboratory testing will commonly reveal high levels of uroporphyrinogen in the urine, clinically referred to as uroporphyrinogenuria. Additionally, testing for common risk factors such as Hepatitis C and hemochromatosis is strongly suggested, as their high prevalence in patients with PCT may require additional treatment. If clinical appearance of PCT is present, but laboratories are negative, one needs to seriously consider the diagnosis of pseudoporphyria.
Type I porphyria cutanea tarda, the sporadic form, is indicated by UROD deficiency that is observed only in hepatic cells and nowhere else in the body. Genetically, these individuals will not exhibit deficiency in the UROD gene, although other genetic factors such as HFE deficiency (resulting in hemochromatosis and the buildup of iron in the liver) are thought to play a key role. Typically in these individuals, a variety of risk factors such as alcohol abuse and Hepatitis C infection conspire to result in the clinical manifestation of PCT.
Patients exhibiting Type II PCT have a specific deficiency in the UROD gene, passed down in an autosomal dominant pattern. Those possessing this deficiency are heterozygous for the UROD gene. They do not show a complete lack of functional uroporphyrinogen decarboxylase, only a deficient form of the enzyme that is marked by reduced conversion of uroporphyrinogen to coproporphyrinogen. Therefore, the expression of uroporphyrinogen decarboxylase will be reduced throughout the body of these individuals, while it is isolated to the liver in Type I patients. While this genetic deficiency is the main distinction between Type I and Type II PCT, the risk factors mentioned before are often seen in patients presenting with Type II PCT. In fact, many people who possess the deficient UROD gene often go their entire lives without having a clinical manifestation of PCT symptoms.
Type III
-
The least common is Type III, which is no different from Type I insofar as the patients possess normal UROD genes. Despite this, Type III PCT is observed in more than one family member, indicating a genetic component unrelated to the expression of uroporphyrinogen decarboxylase.
One study used 74% as the cutoff for UROD activity, with those patients under that number being classified as type II, and those above classified as type III if there was a family history, and type I if there was not.[20]
Genetic variants associated with hemochromatosis have been observed in PCT patients,[9] which may help explain inherited PCT not associated with UROD.
Since PCT is a chronic condition, a comprehensive management of the disease is the most effective means of treatment. Primarily, it is key that patients diagnosed with PCT avoid alcohol consumption, iron supplements, excess exposure to sunlight (especially in the summer), as well as estrogen and chlorinated cyclic hydrocarbons, all of which can potentially exacerbate the disorder. Additionally, the management of excess iron (due to the commonality of hemochromatosis in PCT patients) can be achieved through phlebotomy, whereby blood is systematically drained from the patient. A borderline iron deficiency has been found to have a protective affect by limiting haem synthesis. In the absence of iron, which is to be incorporated in the porphyrin formed in the last step of the synthesis, the mRNA of erythroid 5-aminolevulinate synthase (ALAS-2) is blocked by attachment of an iron-responsive element (IRE) binding cytosolic protein, and transcription of this key enzyme is inhibited.[21]
Low doses of antimalarials can be used.[22] Orally ingested chloroquine is completely absorbed in the gut and is preferentially concentrated in the liver, spleen, and kidneys.[23] They work by removing excess porphyrins from the liver via increasing the excretion rate by forming a coordination complex with the iron center of the porphyrin as well as an intramolecular hydrogen bond between a propionate side chain of the porphyrin and the protonated quinuclidine nitrogen atom of either alkaloid.[24] Due to the presence of the chlorine atom, the entire complex is more water soluble allowing the kidneys to preferentially remove it from the blood stream and expel it through urination.[23][25][26] It should be noted that chloroquine treatment can induce porphyria attacks within the first couple of months of treatment due to the mass mobilization of porphyrins from the liver into the blood stream.[23] Complete remission can be seen within 612 months as each dose of antimalarial can only remove a finite amount of porphyrins and there are generally decades of accumulation to be cleared. Originally, higher doses were used to treat the condition but are no longer recommended because of liver toxicity.[27][28][29] Finally, due to the strong association between PCT and Hepatitis C, the treatment of Hepatitis C (if present) is vital to the effective treatment of PCT. Chloroquine, hydroxychloroquine, and venesection are typically employed in the management strategy.[30]
Porphyria cutanea tarda has a prevalence estimated at approximately 1 in 10,000.[31] An estimated 80% of porphyria cutanea tarda cases are sporadic. The exact frequency is not clear because many people with the condition never experience symptoms and those that do are often misdiagnosed with anything ranging from idiopathic photodermatitis and seasonal allergies to hives.
Porphyria cutanea tarda is implicated in the origin of vampire myths. This is because people with the disease tend to avoid the sun due to photosensitivity (and therefore have a young complexion and pallor) and require blood transfusions. Hypertrichosis (i.e. A full head of hair) will also give a young appearance.
Porphyria Cutanea Tarda is the name of a song by the rock band AFI on their fourth album Black Sails in the Sunset, released on May 18, 1999.
Jump upDi Padova, C.; Marchesi, L.; Cainelli, T.; Gori, G.; Podenzani, S.A.; Rovagnati, P.; Rizzardini, M.; Cantoni, L. (1983). "Effects of Phlebotomy on Urinary Porphyrin Pattern and Liver Histology in Patients with Porphyria Cutanea Tarda". The American Journal of the Medical Sciences. 285 (1): 212. doi:10.1097/00000441-198301000-00001. PMID6824014.
Jump upGoljan, E. F. (2011). Pathology (3rd ed., rev. reprint.). Philadelphia, PA: Mosby/Elsevier.[page needed]
Jump upJackson, A. H.; Ferramola, A. M.; Sancovich, H. A.; Evans, N; Matlin, S. A.; Ryder, D. J.; Smith, S. G. (1976). "Hepta- and hexa-carboxylic porphyrinogen intermediates in haem biosynthesis". Annals of clinical research. 8 Suppl 17: 649. PMID1008499.
Jump upMiller, Dennis M.; Woods, James S. (1993). "Urinary porphyrins as biological indicators of oxidative stress in the kidney". Biochemical Pharmacology. 46 (12): 223541. doi:10.1016/0006-2952(93)90614-3. PMID8274157.
Jump upBrun, Atle; Sandberg, Sverre (1991). "Mechanisms of photosensitivity in porphyric patients with special emphasis on erythropoietic protoporphyria". Journal of Photochemistry and Photobiology B: Biology. 10 (4): 285302. doi:10.1016/1011-1344(91)80015-A. PMID1791486.
Jump upLim, H. W. (1989). "Mechanisms of phototoxicity in porphyria cutanea tarda and erythropoietic protoporphyria". Immunology series. 46: 67185. PMID2488874.
Jump upHeld, H. (2009). "Effect of Alcohol on the Heme and Porphyrin Synthesis Interaction with Phenobarbital and Pyrazole". Digestion. 15 (2): 13646. doi:10.1159/000197995. PMID838185.
Jump upThunell, S (2000). "Porphyrins, porphyrin metabolism and porphyrias. I. Update". Scandinavian journal of clinical and laboratory investigation. 60 (7): 50940. PMID11202048.
Jump upDe Villiers, Katherine A.; Gildenhuys, Johandie; Le Roex, Tanya (2012). "Iron(III) Protoporphyrin IX Complexes of the Antimalarial CinchonaAlkaloids Quinine and Quinidine". ACS Chemical Biology. 7 (4): 66671. doi:10.1021/cb200528z. PMID22276975.
Jump upAsghari-Khiavi, Mehdi; Vongsvivut, Jitraporn; Perepichka, Inna; Mechler, Adam; Wood, Bayden R.; McNaughton, Don; Bohle, D. Scott (2011). "Interaction of quinoline antimalarial drugs with ferriprotoporphyrin IX, a solid state spectroscopy study". Journal of Inorganic Biochemistry. 105 (12): 16629. doi:10.1016/j.jinorgbio.2011.08.005. PMID22079977.
Jump upScholnick, Perry L.; Epstein, John; Marver, Harvey S. (1973). "The Molecular Basis of the Action of Chloroquine in Porphyria Cutanea Tarda". Journal of Investigative Dermatology. 61 (4): 22632. doi:10.1111/1523-1747.ep12676478. PMID4744026.
Based on the agenda, we will be welcoming live questions at designated points during the meeting via live chat. Please comment/provide your questions in the chat section on the webcast. If you are unable to provide question(s) there, please email your question(s) to Jessica at Jessica@porphyriafoundation.org.
To view the meeting live, we will have a webcast streaming in real time. Here is the link: https://youtu.be/AX20OvotxHA
We have attached the link for the PFDD Meeting Agenda for your re-view. Here is the link:
If you are planning to speak at the FDA Meeting, please submit your speech to the APF TODAY to lyonapf@aol.com. You are welcome to say whatever you want, however, we need everyones story sent in order to know where to place you within the program.
If you are planning to speak at the FDA Meeting, please submit your speech to the APF TODAY to lyonapf@aol.com, so that we can know where to place you within the program!!
If you are planning to speak at the FDA Meeting, please submit your speech to the APF by FRIDAY, so that we can know where to place you within the program!!
The meeting will be webcast live on our YouTube Channel. Here is the link:
**For those of you not attending the meeting in person, you do nothave to register in order to watch the live webcast. **
If you misplace this link, you can also gain access via the APF website. Click on the "Register Now" link on the homepage of the APF website under the FDA announcement and it will bring you right to it!
Note: The chat feature will only be accessible through YouTube Live!
The APF meet and greet will be on February 28th at the College Park Marriott (same place as FDA meeting the next morning). The room will be open for an informal meeting space from 5-6PM and there will be a mandatory preparation meeting for all attendees from 6-7PM. After this meeting, you are welcome to go to dinner, hang out with friends, etc. The hotel has an on-site restaurant, The Common, and advance reservations are recommended. Please let the APF know if you plan on attending the meet and greet and preparation meeting ASAP. We need to make sure the space is large enough to accommodate everyone!
Rare Disease Week on Capitol Hill: February 27, 2017 March 2, 2017
Monday - February 20, 2017 @ 10:49:18
Want your voice heard? Submit your story TODAY!
Rare Disease Week on Capitol Hill: February 27, 2017 March 2, 2017
Rare Disease Week on Capitol Hill brings rare disease community members from across the country together to be educated on federal legislative issues, meet other advocates, and share their unique stories with legislators. Submit your stories TODAY to let Congress know about your experiences with this rare disease; how it has impacted your life and let them know which issues are most important to you.
You can submit your personal perspective http://rareadvocates.org/rdw-old/patientstories/ to be hand-delivered to your Representative and Senators on Lobby Day. The deadline to submit perspectives is Sunday, February 19th.
The registration to attend in person in closed, however, you can participate remotely by watching the livestream of the Legislative Conference on Tuesday, February 28th, from 9am to 5pm ET. To register for the free live stream register here: http://action.everylifefoundation.org/â?¦/eveâ?¦/common/public/â?¦ A link to the live stream will be emailed to all registrants the morning of the event.
Submit your story TODAY!
"Remember.Research is the key to your cure!"
Clinuvel Pharmaceuticals Ltd
Monday - February 20, 2017 @ 09:30:07
The February 2017 edition of CLINUVEL's newsletter is out today. CEO Dr Philippe Wolgen provides latest company updates and information for 2017.
If you misplace this link, you can also gain access via the APF website. Click on the "Register Now" link on the homepage of the APF website under the FDA announcement and it will bring you right to it!
Note: The chat feature will only be accessible through YouTube Live!
The APF meet and greet will be on February 28th at the College Park Marriott (same place as FDA meeting the next morning). The room will be open for an informal meeting space from 5-6PM and there will be a mandatory preparation meeting for all attendees from 6-7PM. After this meeting, you are welcome to go to dinner, hang out with friends, etc. The hotel has an on-site restaurant, The Common, and advance reservations are recommended. Please let the APF know if you plan on attending the meet and greet and preparation meeting. We need to make sure the space is large enough to accommodate everyone!
Acute Porphyrias By By Herbert L. Bonkovsky, MD, University of North Carolina at Chapel Hill, and University of Connecticut Health Sciences Center ; Vinaya Maddukuri, MD, Carolinas Medical Center
Wednesday - February 15, 2017 @ 09:30:00
Acute Porphyrias
By Herbert L. Bonkovsky, MD, University of North Carolina at Chapel Hill, and University of Connecticut Health Sciences Center;Vinaya Maddukuri, MD, Carolinas Medical Center
Acute porphyrias result from deficiency of certain enzymes in the heme biosynthetic pathway, resulting in accumulation of heme precursors that cause intermittent attacks of abdominal pain and neurologic symptoms. Attacks are precipitated by certain drugs and other factors. Diagnosis is based on elevated levels of the porphyrin precursors δ-aminolevulinic acid and porphobilinogen in the urine during attacks. Attacks are treated with glucose or, if more severe, IV heme. Symptomatic treatment, including analgesia, is given as necessary.
Patients with VP and HCP, with or without neurovisceral symptoms, may develop bullous eruptions especially on the hands, forearms, face, neck, or other areas of the skin exposed to sunlight.
Among heterozygotes, acute porphyrias are rarely expressed clinically before puberty; after puberty, they are expressed in only about 2 to 4%. Among homozygotes and compound heterozygotes, onset typically is in childhood, and symptoms are often severe.
Precipitating Factors
Many precipitating factors exist, typically accelerating heme biosynthesis above the catalytic capacity of the defective enzyme. Accumulation of the porphyrin precursors porphobilinogen (PBG) and δ-aminolevulinic acid (ALA), or in the case of ALAD-deficiency porphyria, ALA alone, results.
Attacks probably result from several, sometimes unidentifiable, factors. Identified precipitating factors include
Hormonal changes in women
Drugs
Low-calorie, low-carbohydrate diets
Alcohol
Exposure to organic solvents
Infections and other illnesses
Surgery
Emotional stress
Hormonal factors are important. Women are more prone to attacks than men, particularly during periods of hormonal change (eg, luteal phase of the menstrual cycle, during oral contraceptive use, during early weeks of gestation, in the immediate postpartum period). Nevertheless, pregnancy is not contraindicated.
Other factors include drugs (including barbiturates, hydantoins, other antiepileptic drugs, and sulfonamide antibioticssee Table: Drugs and Porphyria*) and reproductive hormones (progesterone and related steroids), particularly those that induce hepatic ALA synthase and cytochrome P-450 enzymes. Attacks usually occur within 24 h after exposure to a precipitating drug.
Exposure to sunlight precipitates cutaneous symptoms in VP and HCP.
Drugs and Porphyria*
Category/Disorder Treated
Unsafe
Safe
Probably Safe
Analgesic
Dextropropoxypheneâ?
Diclofenac
Meprobamate
Propoxyphene
Tramadol
Aspirin
Buprenorphine
Caffeine
Codeine
Morphine
Propofol
Atropine
Dexibuprofenâ?
Fentanyl
Hydromorphone
Ketobemidoneâ?
Ketoprofen
Naproxen
Anesthetic (local)
Lidocaine
Bupivacaine
Articaine
Anesthetic (premedication, induction, or maintenance)
Barbiturates
Atropine
Morphine
Propofol
Alfentanil
Desflurane
Droperidol
Fentanyl
Isoflurane
Remifentanil
Scopolamine
Sufentanil
Antidepressant
Lithium
Fluoxetine
Antidiarrheal
Active carbon
Loperamide
Antiemetic
Chlorpromazine
Granisetron
Ondansetron
Scopolamine
Tropisetronâ?
Anticonvulsant
Barbiturates
Carbamazepine
Diones (paramethadioneâ? , trimethadione)
Felbamate
Lamotrigine
Mephenytoinâ?
Phenytoin
Primidone
Succinimides (ethosuximide, methsuximide)
Valproate
Clonazepam
Diazepam (active seizure)
Gabapentin
Levetiracetam
Topiramate
Vigabatrin
Antihyperglycemic
Sulfonylureas
Acarbose
Insulin
Metformin
Anti-infective
Chloramphenicol
Clindamycin
Erythromycin
Indinavir
Ketoconazole
Mecillinamâ?
Nitrofurantoin
Pivampicillinâ?
Pivmecillinamâ?
Rifampin
Ritonavir
Sulfonamides
Trimethoprim
Acyclovir
Amikacin
Amoxicillin
Amoxicillin with a β-lactamase inhibitor
Ampicillin
Cloxacillinâ?
Dicloxacillin
Fusidic acidâ?
Ganciclovir
Gentamicin
Immune globulin
Immune sera
Methenaminehippurate
Netilmicinâ?
Oseltamivir
Penicillin G
Penicillin V
Piperacillin
Teicoplaninâ?
Tobramycin
Vaccines
Valacyclovir
Vancomycin
Zanamivir
Amphotericin B
Azithromycin
Bacampicillinâ?
Cephalosporins
Ciprofloxacin
Didanosine
Ethambutol
Ertapenem
Famciclovir
Flucytosine
Foscarnet
Fosfomycin
Imipenem/cilastatin
Levofloxacin
Meropenem
Moxifloxacin
Norfloxacinâ?
Ofloxacin
Piperacillin with tazobactam
Ribavirin
Anti-inflammatory or antirheumatic
Hyaluronic acid
Penicillamine
Salicylates
Abacavir
Dexibuprofenâ?
Ibuprofen
Ketoprofen
Lamivudine
Lornoxicamâ?
Naproxen
Piroxicam
Tenofovir disoproxil fumarate
Tenoxicamâ?
Zalcitabineâ?
Anxiolytic, sedative-hypnotic, or antipsychotic
Ethchlorvynolâ?
Glutethimideâ?
Hydroxyzine
Meprobamate
Chlorpromazine
Droperidol
Fluoxetine
Fluphenazine
Haloperidol
Levomepromazineâ?
Prochlorperazine
Propiomazineâ?
Alprazolam
Clozapine
Dixyrazineâ?
Eszopiclone
Lorazepam
Olanzapine
Oxazepam
Perphenazine
Triazolam
Cardiovascular disorders
Dihydralazineâ?
Ergoloid mesylate
Hydralazine
Lidocaine
Methyldopa
Nifedipine
Spironolactone
Amiloride
β-Blockers
Cholestyramine
Colestipol
Digitalis glycosides
Diltiazem
Enalapril
Epinephrine
Heparins
Lisinopril
Losartan
Niacin
Organic nitrates
Adenosine
Amrinone
Bendroflumethiazide
Bezafibrateâ?
Bumetanide
Digoxin
Dobutamine
Dopamine
Dopexamineâ?
Doxazosin
Ethacrynic acid
Etilefrineâ?
Fenofibrate
Furosemide
Hydrochlorothiazide
Milrinone
Phenylephrine
Prostaglandins
Quinidine
Hormones
Danazol
Progesterone
Synthetic progestins
Nonreproductive hormones, including glucocorticoids
Natural estrogens
Laxatives
Bisacodyl
Cascara sagrada
Dietary fiber
Lactitolâ?
Lactulose
Lauryl sulfate
Psyllium seed
Senna glycosides
Sodium docusate
Sodium picosulfateâ?
Sorbitol
Migraines
Ergots
Muscle relaxants
Carisoprodol
Orphenadrine
Atracurium
Cisatracurium
Mivacuriumâ?
Pancuronium
Rocuronium
Succinylcholine(suxamethonium)
Vecuronium
Baclofen
Osteoporosis
Bisphosphonates
Calcium supplements
Peptic ulcers
Alginic acid
Ca-containing antacids
Cimetidine
Mg-containing antacids
Sucralfate
Famotidine
Misoprostol
Nizatidine
Ranitidine
Respiratory disorders
Clemastine
Dimenhydrinate
Albuterol (salbutamol)
Alimemazineâ?
Codeine
Corticosteroids
Dipalmitoyl phosphatidylcholine
Dornase alfa
Ephedrine
Ethylmorphine
Ipratropium
Phenylpropanolamineâ?
Phospholipid surfactant
Bambuterolâ?
Cromolyn
Desloratadine
Fenoterolâ?
Fexofenadine
Formoterol
Levocabastineâ?
Lidocaine (solution for gargling)
Loratadine
Mizolastineâ?
Oxymetazoline
Salmeterol
Terbutaline
Tiotropium
*The classification of the drugs in the list is based on a combination of clinical observations, case reports in the literature, and theoretical considerations derived from the structure and metabolism of the substances. However, clinical observations may in many cases be unreliable. Also, the biochemical and molecular-biologic models for the activation of the disease are incomplete. This list is meant as guidance only and is neither complete nor applicable to all patients. Drugs must always be used cautiously in people who carry genes for acute porphyria. For questions about specific drugs, physicians can consult www.drugs-porphyria.org.
â? Not available in the US.
Symptoms and Signs
Symptoms and signs of acute porphyrias involve the nervous system, abdomen, or both (neurovisceral). Attacks develop over hours or days and can last up to several weeks. Most gene carriers experience no, or only a few, attacks during their lifetime. Others experience recurrent symptoms. In women, recurrent attacks often coincide with the luteal phase of the menstrual cycle.
The acute porphyric attack
Constipation, fatigue, irritability, and insomnia typically precede an acute attack. The most common symptoms of an attack are abdominal pain and vomiting. The pain may be excruciating and is disproportionate to abdominal tenderness or other physical signs. Abdominal manifestations may result from effects on visceral nerves or from local vasoconstrictive ischemia. Because there is no inflammation, the abdomen is not tender and there are no peritoneal signs. Temperature and WBC count are normal or only slightly increased. Bowel distention may develop as a result of paralytic ileus. The urine is red or reddish brown and positive for PBG during an attack.
All components of the peripheral nervous system and the CNS may be involved. Motor neuropathy is common with severe and prolonged attacks. Muscle weakness usually begins in the extremities but can involve any motor neuron or cranial nerve and proceed to tetraplegia. Bulbar involvement can cause respiratory failure.
CNS involvement may cause seizures or mental disturbances (eg, apathy, depression, agitation, frank psychosis, hallucinations). Seizures, psychotic behavior, and hallucinations may be due to or exacerbated by hyponatremia or hypomagnesemia, which can also contribute to cardiac arrhythmias. Hyponatremia may occur during an acute attack due to excessive vasopressin (antidiuretic hormone [ADH]) release and/or administration of hypotonic IV solutions (5% or 10% dextrose in water), a standard therapy for acute attacks.
Excess catecholamines generally cause restlessness and tachycardia. Rarely, catecholamine-induced arrhythmias cause sudden death. Labile hypertension with transiently high BP may cause vascular changes progressing to irreversible hypertension if untreated. Renal failure in acute porphyria is multifactorial; acute hypertension (possibly leading to chronic hypertension) is likely a main precipitating factor.
Subacute or subchronic symptoms
Some patients have prolonged symptoms of lesser intensity (eg, obstipation, fatigue, headache, back or thigh pain, paresthesia, tachycardia, dyspnea, insomnia, depression, anxiety or other disturbances of mood, seizures).
Skin symptoms in VP and HCP
Fragile skin and bullous eruptions may develop on sun-exposed areas, even in the absence of neurovisceral symptoms. Often patients are not aware of the connection to sun exposure. Cutaneous manifestations are identical to those of porphyria cutanea tarda; lesions typically occur on the dorsal aspects of the hands and forearms, the face, ears, and neck.
Late manifestations of acute porphyrias
Motor involvement during acute attacks may lead to persistent muscle weakness and muscle atrophy between attacks. Cirrhosis, hepatocellular carcinoma, systemic arterial hypertension, and renal impairment become more common after middle age in AIP and possibly also in VP and HCP, especially in patients with previous porphyric attacks.
Diagnosis
Urine screen for PBG
If urine results are positive, quantitative ALA and PBG determination
For confirmation of AIP, measurement of PBG deaminase activity in erythrocytes
Genetic analysis if type is to be identified
Acute attack
Misdiagnosis is common because the acute attack is confused with other causes of acute abdomen (sometimes leading to unnecessary surgery) or with a primary neurologic or mental disorder. However, in patients previously diagnosed as gene carriers or who have a positive family history, porphyria should be suspected. Still, even in known gene carriers, other causes must be considered.
Red or reddish brown urine, not present before onset of symptoms, is a cardinal sign and is present during full-blown attacks. A urine specimen should be examined in patients with abdominal pain of unknown cause, especially if severe constipation, vomiting, tachycardia, muscle weakness, bulbar involvement, or mental symptoms occur.
If porphyria is suspected, the urine is analyzed for PBG using a rapid qualitative or semiquantitative determination. A positive result or high clinical suspicion necessitates quantitative ALA and PBG measurements preferentially obtained from the same specimen. PBG and ALA levels > 5 times normal indicate an acute porphyric attack unless patients are gene carriers in whom porphyrin precursor excretion occurs at similar levels even during the latent phase of the disorder.
If urinary PBG and ALA levels are normal, an alternative diagnosis must be considered. Measurement of urinary total porphyrins and high-performance liquid chromatography profiles of these porphyrins are helpful. Elevated urinary ALA and coproporphyrin with normal or slightly increased PBG suggests lead poisoning, ALAD-deficiency porphyria, or hereditary tyrosinemia type 1. Analysis of a 24-h urine specimen is not necessary. Instead, a random urine specimen is used, and PBG and ALA levels are corrected for dilution by relating to the creatinine level of the sample. Electrolytes and Mg should be measured. Hyponatremia may be present because of excessive vomiting or diarrhea after hypotonic fluid replacement or because of the syndrome of inappropriate antidiuretic hormone secretion (SIADH).
Determination of acute porphyria type
Because treatment does not depend on the type of acute porphyria, identification of the specific type is valuable mainly for finding gene carriers among relatives. When the type and mutation are already known from previous testing of relatives, the diagnosis is clear but may be confirmed by gene analysis. Activity of the enzymes ALAD and PBGD in the red blood cells is readily measurable and can be helpful for establishing the diagnosis in ALAD-deficiency porphyria and acute intermittent porphyria, respectively. RBC PBG deaminase levels that are about 50% of normal suggest AIP. If there is no family history to guide the diagnosis, the different forms of acute porphyria are distinguished by characteristic patterns of porphyrin (and precursor) accumulation and excretion in plasma, urine, and stool. When urinalysis reveals increased levels of ALA and PBG, fecal porphyrins may be measured. Fecal porphyrins are usually normal or minimally increased in AIP but elevated in HCP and VP. Often, these markers are not present in the quiescent phase of the disorder. Plasma fluorescence emission after excitation with Soret band of light (~410 nm) can be used to differentiate HCP and VP, which have different peak emissions.
Family studies in acute porphyrias
Children of a gene carrier for an autosomal dominant form of acute porphyria (AIP, HCP, VP) have a 50% risk of inheriting the disorder. In contrast, children of patients with ALAD-deficiency porphyria (autosomal recessive inheritance) are obligate carriers but are very unlikely to develop clinical disease. Because early diagnosis followed by counseling reduces the risk of morbidity, children in affected families should be tested before the onset of puberty. Genetic testing is used if the mutation has been identified in the index case. If not, pertinent RBC or WBC enzyme levels are measured. Gene analysis can be used for in utero diagnosis (using amniocentesis or chorionic villus sampling) but is seldom indicated because of the favorable outlook for most gene carriers.
Prognosis
Advances in medical care and self-care have improved the prognosis of acute porphyrias for symptomatic patients. Still, some patients develop recurrent crises or progressive disease with permanent paralysis or renal failure. Also, frequent need for opioids analgesics may give rise to opioid dependence.
Treatment
Triggers eliminated if possible
Dextrose (oral or IV)
IV heme
Treatment of the acute attack is identical for all the acute porphyrias. Possible triggers (eg, excessive alcohol use, drugs) are identified and eliminated. Unless the attack is mild, patients are hospitalized in a darkened, quiet, private room. Heart rate, BP, and fluid and electrolyte balance are monitored. Neurologic status, bladder function, muscle and tendon function, respiratory function, and O2 saturation are continuously monitored. Symptoms (eg, pain, vomiting) are treated with nonporphyrinogenic drugs as needed (see Table: Drugs and Porphyria*).
Dextrose 300 to 500 g daily down-regulates hepatic ALA synthase (ALAS 1) and relieves symptoms. Dextrose can be given by mouth if patients are not vomiting; otherwise, it is given IV. The usual regimen is 3 L of 10% dextrose solution, given by a central venous catheter over 24 h (125 mL/h). However, to avoid overhydration with consequent hyponatremia, 1 L of 50% dextrose solution can be used instead.
IV heme is more effective than dextrose and should be given immediately in severe attacks, electrolyte imbalance, or muscle weakness. Heme usually resolves symptoms in 3 to 4 days. If heme therapy is delayed, nerve damage is more severe and recovery is slower and possibly incomplete. Heme is available in the US as lyophilized hematin to be reconstituted in a glass vial with sterile water. The dose is 3 to 4 mg/kg IV once/day for 4 days. An alternative is heme arginate, which is given at the same dose, except that it is diluted in 5% dextrose or half-normal or quarter-normal saline. Hematin and heme arginate may cause venous thrombosis and/or thrombophlebitis. Risk of these adverse events appears to be lower if the heme is administered bound to human serum albumin. Such binding also decreases the rate of development of hematin aggregates. Thus, most authorities recommend administration of hematin or heme arginate with human serum albumin.
Recurrent attacks
In patients with severe recurrent attacks, who are at risk of renal damage or permanent neurologic damage, liver transplantation is an option. Successful liver transplantation leads to permanent cure of all acute porphyrias other than ALAD-deficiency porphyria. Patients with acute porphyrias should not serve as liver donors even though their liver may appear structurally normal (ie, no cirrhosis) because recipients have developed acute porphyric syndromes; such an outcome helped establish that the acute porphyrias are hepatic disorders. Renal transplantation, with or without simultaneous liver exchange, should be considered in patients with active disease and terminal renal failure because there is considerable risk that nerve damage will progress at the start of dialysis.
Prevention
Carriers of acute porphyria should avoid the following:
Exposure to organic solvents (eg, in painting or dry cleaning)
Crash diets
Periods of starvation
Diets for obesity should provide gradual weight loss and be adopted only during periods of remission. Carriers of VP or HCP should minimize sun exposure; sunscreens that block only ultraviolet B light are ineffective, but opaque zinc oxide or titanium dioxide preparations are beneficial. Support associations, such as the American Porphyria Foundation and the European Porphyria Network, can provide written information and direct counseling.
Patients should be identified prominently in the medical record as carriers and should carry a card verifying the carrier state and precautions to be observed.
A high-carbohydrate diet may decrease the risk of acute attacks. Some patients can sometimes treat mild acute attacks by increasing their intake of dextrose or glucose. Prolonged use should be avoided in order to decrease risk of obesity and dental caries.
Patients who experience recurrent and predictable attacks (typically women with attacks related to the menstrual cycle) may benefit from prophylactic heme therapy given shortly before the expected onset. There is no standardized regimen; a specialist should be consulted. Frequent premenstrual attacks in some women are aborted by administration of a gonadotropin-releasing hormone agonist plus low-dose estrogen. Low-dose oral contraceptives are sometimes used successfully, but the progestin component is likely to exacerbate the porphyria.
To prevent renal damage, chronic hypertension should be treated aggressively (using safe drugs). Patients with evidence of impaired renal function are referred to a nephrologist. Recent anecdotal experience indicates that tolvaptan, an ADH blocker, is helpful in the management of hyponatremia during acute attacks.
The incidence of hepatocellular cancer is high among carriers of acute porphyria, especially in patients with active disease. Patients who are > 50 should undergo yearly or twice yearly surveillance, including liver screening with ultrasonography. Early intervention can be curative and increases life expectancy.
Key Points
Acute porphyrias cause intermittent attacks of abdominal pain and neurologic symptoms; some types also have cutaneous manifestations that are triggered by sun exposure.
Attacks have many triggers, including hormones, drugs, low-calorie and low-carbohydrate diets, and alcohol ingestion.
Attacks typically involve severe abdominal pain (with a non-tender abdomen) and vomiting; any component of the peripheral and central nervous system may be affected but muscle weakness is common.
Urine is often reddish-brown during an attack.
Do a qualitative urine test for PBG and confirm a positive result with quantitative ALA and PBG measurements
Treat acute attacks with oral or IV dextrose and, for severe attacks, IV heme.
If you misplace this link, you can also gain access via the APF website. Click on the "Register Now" link on the homepage of the APF website under the FDA announcement and it will bring you right to it!
Note: The chat feature will only be accessible through YouTube Live!
The APF meet and greet will be on February 28th at the College Park Marriott (same place as FDA meeting the next morning). The room will be open for an informal meeting space from 5-6PM and there will be a mandatory preparation meeting for all attendees from 6-7PM. After this meeting, you are welcome to go to dinner, hang out with friends, etc. The hotel has an on-site restaurant, The Common, and advance reservations are recommended. Please let the APF know if you plan on attending the meet and greet and preparation meeting. We need to make sure the space is large enough to accommodate everyone!
My name Brigitta Owen. My story begins with my grandmother who died at the age of 27 just 10 days after giving birth to my father. She died of colic after eating too many cherries. I know now that she had an acute porphyria but then, they just didn't know better!
When I came into teenage years, I could not go out in the sun without a hat or some protection. I always ended up being sick and suffering from what I thought was "sunstroke." I did not start having real attacks until I got pregnant the first time at the age of 19. I got pregnant and miscarried 7 times and each time my symptoms got worse. Terrible pain, changes in my personality, was put down to being afraid of another miscarriage. I always ended up in a psych ward where they gave me medicine that made me worse.
When I fainted, I got smacked and was told to pull myself together. I was black and blue, but they really thought I was putting it on! When I wasn't sent to the psych ward, they opened me up first and never found anything and then sent me to psych hospital.
My 8th pregnancy lasted 28 weeks. I gave birth by cesarean section to my first daughter, Michelle. She did well for 2 weeks but got an infection she couldn't fight being so small. Sadly, we lost her at just 3 weeks old. My symptoms were severe and took a long time to subside. We decided to try one more time if it did not work we would stop. I got pregnant very quickly and, of course, I ended up in hospital. My instinct told me something was wrong, but even my husband believed it was all in my head.
I was born in Switzerland but lived in the UK (Wales) since getting married. I was desperate and phoned my mum and asked if I could come home as nobody believed me. My husband drove me to London where I was carried on to the plane. The pilot came to ask what was wrong, I just said I was 9 weeks pregnant and wanted to go home. Thank God he didn't send me away!!! When I got home, the first thing my mum did is to take me to hospital. I only agreed if she took me home again. She had to promise as I thought they would not believe me anyway! They took a blood and urine sample. My mum took me home. Early in the morning, the hospital phoned and said for her to bring me back to hospital as they knew what I had. They left my urine standing and it turned purple! Finally, I had a diagnosis after 7 long years to hell and back!! I was suffering from Acute porphyria. The doctor said if I waited another week I would not be writing to you now!!
I stayed in Switzerland in hospital 3 month before I was well enough to go home to Wales. I was hospitalized nearly all my pregnancy. I gave birth to my miracle Heidi, who was born healthy all be it 6 weeks early. Straight after birth I was sterilized. I wanted my husband to have the operation, which he refused. Even though it meant me having a risky operation and anesthetic. My Porphyria was very active now. I had flare-ups with every hormone change .I haven't been drinking a lot of alcohol before and now I don't drink at all. I am very careful with medicine.
I was admitted to hospital a lot. They were terrible experiences. They didn't believe I was in a lot of pain. I was injected with water just to see if I was lying. They put me in a side ward and took the bell away. I know I am really difficult when I am in a flare-up. My husband couldn't cope anymore. He was getting so angry every time he could see I was unwell again. Slamming doors, even leaving. This just made me worse.
I ended up in hospital again so upset that he left me. All the nurse said that if she had a wife like me she would have left a long time ago. I felt so alone and guilty.
I understood my husband as well. Its not easy to have a wife who is always ill. The whole situation was hell for both of us. We decided to go our separate ways. I went back home to Switzerland. Its the best thing I ever did. My doctor is fantastic. He changed my life. First of all he believed me. I got a port. He registered me as a chronic pain patient. This enables me to get pain relief. I don't get stressed anymore. I am well enough to work full time. Its not all rosy if I end up in hospital, I still get doctors who won't believe my pain. As soon as I sense disbelief, it catapults me back to those years of hell, and I feel I have to fight for my life.
My daughter Heidi, who is 30 years old, went through genetic testing and is positive. Until now she is well. I hope and pray it stays that way!! Since being able to share storys and have the privilege to meet other porphyria sufferers from all around the globe and finding out about the American porphyria foundation, it gives me hope for the future. Not just for me but also my daughter and grandchildren.
I'd like to thank you for listening to my story even though it was a struggle Im not sorry!! It made me to the person I am today with empathy and compassion which we need a lot more of in this day and age.
If you misplace this link, you can also gain access via the APF website. Click on the "Register Now" link on the homepage of the APF website under the FDA announcement and it will bring you right to it!
Note: The chat feature will only be accessible through YouTube Live!
The APF meet and greet will be on February 28th at the College Park Marriott (same place as FDA meeting the next morning). The room will be open for an informal meeting space from 5-6PM and there will be a mandatory preparation meeting for all attendees from 6-7PM. After this meeting, you are welcome to go to dinner, hang out with friends, etc. The hotel has an on-site restaurant, The Common, and advance reservations are recommended. Please let the APF know if you plan on attending the meet and greet and preparation meeting. We need to make sure the space is large enough to accommodate everyone!
Janie Williams: My Story I always felt my personal story was so private that it was my secret. However, it is now time to tell it in hopes it will help others. I was raised to have a passion for life and that anything you can dream you can achieve, and also, if you werent well enough to go to school, you had to stay in bed. As a young child, I always had abdominal pain and stomach aches. We just referred to it as having an Arrington stomach (named after my maiden name because my Dad and his bothers had stomach problems all their lives). No matter how many times I would tell doctors, they would say it was nothing and it would not shorten my life. It finally got to the point where sometimes the abdominal pain was so fierce I wondered if I should actually pray for death. I would go to the hospital, only to be sent home saying I just had a low grade infection and to go to my doctor the next day. And, of course, the next day the doctor would tell me the same thing. In my 20s, being single and along with not knowing I had Porphyria, I thoroughly enjoyed the beach sun and, of course, engaged in social drinking which are big no-nos for one with this disease. At age 29, after enjoying the summer sun, my urine was the color of dark, brick red. I had blisters on the back of my hands and on the end of my toes. The pain was so fierce I could not even wear shoes. On a hot day, I would feel like my skin was on fire and itched so much that if I could unzip my skin and run away I would. By now, I had been to 11 doctors and decided to try a dermatologist. He took one look at me and said I had a rare genetic liver disease called Porphyria and promptly sent me to the University of Southern California which at that time studied Porphyria at their center for rare diseases. To treat this condition, I had one pint of blood removed every other week for the next four years. I obviously learned how to avoid these attacks by eliminating alcohol and using very smart judgment about the sun during the hottest months which include wearing sun protective clothing and playing golf very early in the morning. As the saying goes, a little knowledge can be dangerous. In 1994, at age 52, I had severe abdominal pain and went to the hospital. At that time about 25% of Porphyria people who went to the hospital died because they were given the wrong medication. So, of course I told them I had Porphyria, however, I knew this was different and that I was not having an attack. They said I just had a low grade infection and sent me home. The second hospital also tried to send me home and fortunately I refused to leave! The bottom line here is, I ended up with a 4 day old ruptured appendix with gangrene, peritonitis, and abscesses. Plus, after seven weeks, I ended back in the hospital for 10 days with bleeding adhesions. When people ask me how long it took me to recover, I say physically and emotionally, 10 years! I became a Doctor of Nutrition to save my own life, however, it is through the American Porphyria Foundation that I have received the most education to understand this disease. For a person with this disease, the quality of ones life is being able understand what is going on and to be able to avoid attacks. Right now, I am living my American Dream of feeling as GREAT as possible everyday and enjoying life to the fullest. This is definitely a full time job making sure I do everything possible 24 hours a day to avoid Porphyria attacks. Living with this disease can be very degrading. For me, I have chosen to receive all the education and knowledge I can to live as normal a life as possible because I deserve it, and so does my husband, family and friends.
Thank you Janie for sharing your story. Become your own advocate for Porphyria disease and awareness! If you would like to share your story with the APF please let me know @ Amy.apf@gmail.com
The FDA Meeting for Acute Porphyrias is Weeks Away!
Wednesday - February 1, 2017 @ 09:00:11
The FDA Meeting for Acute Porphyrias is Weeks Away!
The FDA Meeting for Acute Porphyrias is quickly approaching, but there is still time to register! We need as many patients to attend as possible and tell the FDA their experience about porphyria, treatment, critical needs, etc.
Please let Jessica at the APF know that you are planning to attend. She can be reached by email at Jessica@porphyriafoundation.org or by phone at the APF office at 866-APF-3635 or 713-266-9617. There are also travel scholarships available for those that need financial assistance in order to attend this important meeting. Contact the APF office to apply today!
If you are still interested in reserving a hotel room in the APF room block, the deadline is MONDAY, FEBRUARY 6th. There are a limited number of rooms available, and they are first come, first serve. Contact the APF office for instructions on how to reserve your room.
Please do not hesitate to contact us if you have ANY questions surrounding this important event.
Dear Friends, The APF will be hosting a Patient Focused Drug Development (PFDD) meeting for the Acute Porphyrias on Wednesday, March 1, 2017 at theâ?¦
EVENTBRITE.COM
Partner Up whith The Mighty! Learn more
Friday - January 27, 2017 @ 09:30:26
We're Partnering With The Mighty!
We're thrilled to announce a new partnership that will bring our resources in front of The Mighty's wide-reaching readership. We will now have a growing home page on The Mighty and appear on many stories on the site.
The Mighty is a story-based health community focused on improving the lives of people facing disease, disorder, mental illness and disability. More than half of Americans are facing serious health conditions or medical issues. They want more than information. They want to be inspired. The Mighty publishes real stories about real people facing real challenges.
We're dedicated to helping people with porphyria in their lives. With this partnership, we'll be able to help even more people.
We encourage you to submit a story to The Mighty and make your voice heard.
Welcome to our Mighty community!
Were so glad youre here.
By subscribing to our newsletter, youll receive a weekly digest of compelling personal essays and the latest news stories about disability, disease and mental illness. We strive to create and deliver the best resources for our readers. Well never send you spam, and you can unsubscribe anytime (but we hope you dont!).
Youll also be introduced to our growing Mighty contributor community the people behind many of the powerful stories we highlight every week. Hopefully youll be inspired to share a story of your own, too. (Read our submissions page for more info on that.)
Stay tuned for our next edition. We can't wait for you to read it.
Sincerely,
The Mighty Team
P.S.: We have more newsletter topics, too! Check out our growing list of newsletters to make sure youre staying up to date on the topics that matter most to you.
Researcher Story: Why Participate in Clinical Research?
NIH-supported ResearchMatch.org helps volunteers and researchers connect for clinical trials. Researchers and clinical trial participants explain what it's like to volunteer for a trial and how it promotes medical advances.
What are clinical trials and why do people participate?
Clinical trials are part of clinical research and at the heart of all medical advances. Clinical trials look at new ways to prevent, detect, or treat disease. Treatments might be new drugs or new combinations of drugs, new surgical procedures or devices, or new ways to use existing treatments. The goal of clinical trials is to determine if a new test or treatment works and is safe. Clinical trials can also look at other aspects of care, such as improving the quality of life for people with chronic illnesses.
People participate in clinical trials for a variety of reasons. Healthy volunteers say they participate to help others and to contribute to moving science forward. Participants with an illness or disease also participate to help others, but also to possibly receive the newest treatment and to have the additional care and attention from the clinical trial staff. Clinical trials offer hope for many people and an opportunity to help researchers find better treatments for others in the future.
What is clinical research?
Clinical research is medical research that involves people like you. People volunteer to participate in carefully conducted investigations that ultimately uncover better ways to treat, prevent, diagnose, and understand human disease. Clinical research includes trials that test new treatments and therapies as well as longterm natural history studies, which provide valuable information about how disease and health progress.
The idea
The idea for a clinical research study also known as a clinical trial often originates in the laboratory. After researchers test new therapies or procedures in the laboratory and in animal studies, the most promising experimental treatments are moved into clinical trials, which are conducted in phases. During a trial, more information is gained about an experimental treatment, its risks, and its effectiveness.
The protocol
Clinical research is conducted according to a plan known as a protocol. The protocol is carefully designed to safeguard the participants health and answer specific research questions. A protocol describes the following:
Who is eligible to participate in the trial
Details about tests, procedures, medications, and dosages
The length of the study and what information will be gathered
A clinical study is led by a principal investigator (PI), who is often a doctor. Members of the research team regularly monitor the participants health to determine the studys safety and effectiveness.
IRB review
Most, but not all, clinical trials in the United States are approved and monitored by an Institutional Review Board (IRB) in order to ensure that the risks are minimal and are worth any potential benefits. An IRB is an independent committee that consists of physicians, statisticians, and members of the community who ensure that clinical trials are ethical and that the rights of participants are protected. Potential research participants should ask the sponsor or research coordinator whether the research they are considering participating in was reviewed by an IRB.
Sponsors
Clinical trials are sponsored or funded by various organizations or individuals, including physicians, foundations, medical institutions, voluntary groups, and pharmaceutical companies, as well as federal agencies such as the National Institutes of Health and the Department of Veterans Affairs.
Informed consent
Informed consent is the process of providing potential participants with the key facts about a clinical trial before they decide whether to participate. The process of informed consent (providing additional information) continues throughout the study. To help someone decide whether or not to participate, members of the research team explain the details of the study. Translation or interpretive assistance can be provided for participants with limited English proficiency. The research team provides an informed consent document that includes details about the study, such as its purpose, duration, required procedures, and who to contact for further information. The informed consent document also explains risks and potential benefits. The participant then decides whether to sign the document. Informed consent is not a contract. Volunteers are free to withdraw from the study completely or to refuse particular treatments or tests at any time. Sometimes, however, this will make them ineligible to continue the study.
Types of clinical trials
There are different types of clinical trials.
Natural history studies provide valuable information about how disease and health progress.
Prevention trials look for better ways to prevent a disease in people who have never had the disease or to prevent the disease from returning. Better approaches may include medicines, vaccines, or lifestyle changes, among other things.
Screening trials test the best way to detect certain diseases or health conditions.
Diagnostic trials determine better tests or procedures for diagnosing a particular disease or condition.
Treatment trials test new treatments, new combinations of drugs, or new approaches to surgery or radiation therapy.
Quality of life trials (or supportive care trials) explore and measure ways to improve the comfort and quality of life of people with a chronic illness.
Phases of clinical trials
Clinical trials are conducted in phases. Each phase has a different purpose and helps researchers answer different questions.
Phase I trials: Researchers test an experimental drug or treatment in a small group of people (2080) for the first time. The purpose is to evaluate its safety and identify side effects.
Phase II trials: The experimental drug or treatment is administered to a larger group of people (100300) to determine its effectiveness and to further evaluate its safety.
Phase III trials: The experimental drug or treatment is administered to large groups of people (1,0003,000) to confirm its effectiveness, monitor side effects, compare it with standard or equivalent treatments, and collect information that will allow the experimental drug or treatment to be used safely.
Phase IV trials: After a drug is approved by the FDA and made available to the public, researchers track its safety, seeking more information about a drug or treatments risks, benefits, and optimal use.
Some concepts to understand
Typically, clinical trials compare a new product or therapy with another that already exists to determine if the new one is as successful as, or better than, the existing one. In some studies, participants may be assigned to receive a placebo (an inactive product that resembles the test product, but without its treatment value).
Comparing a new product with a placebo can be the fastest and most reliable way to demonstrate the new products therapeutic effectiveness. However, placebos are not used if a patient would be put at risk particularly in the study of treatments for serious illnesses by not having effective therapy. Most of these studies compare new products with an approved therapy. Potential participants are told if placebos will be used in the study before they enter a trial.
Randomization is the process by which two or more alternative treatments are assigned to volunteers by chance rather than by choice. This is done to avoid any bias with investigators assigning volunteers to one group or another. The results of each treatment are compared at specific points during a trial, which may last for years. When one treatment is found superior, the trial is stopped so that the fewest volunteers receive the less beneficial treatment.
In single-ordouble-blind studies, also called single- or double-masked studies, the participants do not know which medicine is being used, so they can describe what happens without bias. "Blind" (or "masked") studies are designed to prevent members of the research team or study participants from influencing the results. This allows scientifically accurate conclusions. In single-blind ("single-masked") studies, only the patient is not told what is being administered. In a double-blind study, only the pharmacist knows; members of the research team are not told which patients are getting which medication, so that their observations will not be biased. If medically necessary, however, it is always possible to find out what the patient is taking.
Who participates in clinical trials?
Christopher Futcher / iStock
Many different types of people participate in clinical trials. Some are healthy, while others may have illnesses. A healthy volunteer is a person with no known significant health problems who participates in clinical research to test a new drug, device, or intervention. Research procedures with healthy volunteers are designed to develop new knowledge, not to provide direct benefit to study participants. Healthy volunteers have always played an important role in research.
Healthy volunteers are needed for several reasons. When developing a new technique, such as a blood test or imaging device, healthy volunteers (formerly called "normal volunteers") help define the limits of "normal." These volunteers serve as controls for patient groups and are often matched to patients on characteristics such as age, gender, or family relationship. They receive the same test, procedure, or drug the patient group receives. Investigators learn about the disease process by comparing the patient group to the healthy volunteers.
Factors like how much of your time is needed, discomfort you may feel, or risk involved depends on the trial. While some require minimal amounts of time and effort, other studies may require a major commitment in time and effort on behalf of the volunteer, and may involve some discomfort. The research procedure may also carry some risk. The consent process for healthy volunteers includes a detailed discussion of the study's procedures and tests.
A patient volunteer has a known health problem and participates in research to better understand, diagnose, treat, or cure that disease or condition. Research procedures with a patient volunteer help develop new knowledge. These procedures may or may not benefit the study participants.
Patient volunteers may be involved in studies similar to those in which healthy volunteers participate. These studies involve drugs, devices, or interventions designed to prevent, treat, or cure disease. Although these studies may provide direct benefit to patient volunteers, the main aim is to prove, by scientific means, the effects and limitations of the experimental treatment. Consequently, some patients serve as controls by not taking the test drug, or by receiving test doses of the drug large enough only to show that it is present, but not at a level that can treat the condition. A study's benefits may be indirect for the volunteers but may help others.
All clinical trials have guidelines about who can participate, called Inclusion/Exclusion Criteria. Factors that allow someone to participate in a clinical trial are "inclusion criteria." Those that exclude or not allow participation are "exclusion criteria." These criteria are based on factors such as age, gender, the type and stage of a disease, previous treatment history, and other medical conditions. Before joining a clinical trial, a participant must qualify for the study. Some research studies seek participants with illnesses or conditions to be studied in the clinical trial, while others need healthy volunteers.
Some studies need both types. Inclusion and exclusion criteria are not used to reject people personally; rather, the criteria are used to identify appropriate participants and keep them safe, and to help ensure that researchers can find new information they need.
What do I need to know if I am thinking about participating?
Xsandra/iStock
Risks and benefits
Clinical trials involve risks, just as routine medical care and the activities of daily living. When weighing the risks of research, you can consider two important factors:
the degree of harm that could result from participating in the study, and
the chance of any harm occurring.
Most clinical studies pose the risk of minor discomfort, which lasts only a short time. However, some study participants experience complications that require medical attention. In rare cases, participants have been seriously injured or have died of complications resulting from their participation in trials of experimental therapies. The specific risks associated with a research protocol are described in detail in the informed consent document, which participants are asked to sign before participating in research. Also, a member of the research team explains the major risks of participating in a study and will answer any questions you have about the study. Before deciding to participate, carefully consider possible risks and benefits.
Potential benefits
Well-designed and well-executed clinical trials provide the best approach for participants to:
Play an active role in their health care.
Gain access to new research treatments before they are widely available.
Receive regular and careful medical attention from a research team that includes doctors and other health professionals.
Help others by contributing to medical research.
Potential risks
Risks to participating in clinical trials include the following:
There may be unpleasant, serious, or even life-threatening side effects to experimental treatment.
The study may require more time and attention than standard treatment would, including visits to the study site, more blood tests, more treatments, hospital stays, or complex dosage requirements.
What questions should I ask if offered a clinical trial?
If you are offered a clinical trial, feel free to ask any questions or bring up any issues concerning the trial at any time. The following suggestions may give you some ideas as you think about your own questions.
The study
What is the purpose of the study?
Why do researchers think the approach may be effective?
Who will fund the study?
Who has reviewed and approved the study?
How are study results and safety of participants being checked?
How long will the study last?
What will my responsibilities be if I participate?
Possible risks and benefits
What are my possible short-term benefits?
What are my possible long-term benefits?
What are my short-term risks, such as side effects?
What are my possible long-term risks?
What other options do people with my disease have?
How do the possible risks and benefits of this trial compare with those options?
Participation and care
What kinds of therapies, procedures and /or tests will I have during the trial?
Will they hurt, and if so, for how long?
How do the tests in the study compare with those I would have outside of the trial?
Will I be able to take my regular medications while in the clinical trial?
Where will I have my medical care?
Who will be in charge of my care?
Personal issues
How could being in this study affect my daily life?
Can I talk to other people in the study?
Cost issues
Will I have to pay for any part of the trial such as tests or the study drug?
If so, what will the charges likely be?
What is my health insurance likely to cover?
Who can help answer any questions from my insurance company or health plan?
Will there be any travel or child care costs that I need to consider while I am in the trial?
Tips for asking your doctor about trials
Consider taking a family member or friend along, for support and for help in asking questions or recording answers.
Plan ahead what to ask but don't hesitate to ask any new questions you think of while you're there.
Write down your questions in advance, to make sure you remember to ask them all.
Write down the answers, so that you can review them whenever you want.
Ask about bringing a tape recorder to make a taped record of what's said (even if you write down answers).
This information courtesy of Cancer.gov.
How Am I Protected?
Mark Bowden / iStock
Ethical guidelines
The goal of clinical research is to develop knowledge that improves human health or increases understanding of human biology. People who participate in clinical research make it possible for this to occur. The path to finding out if a new drug is safe or effective is to test it on patient volunteers. By placing some people at risk of harm for the good of others, clinical research has the potential to exploit patient volunteers. The purpose of ethical guidelines is both to protect patient volunteers and to preserve the integrity of the science. Ethical guidelines in place today were primarily a response to past research abuses.
Informed consent
Informed consent is the process of learning the key facts about a clinical trial before deciding whether to participate. The process of providing information to participants continues throughout the study. To help someone decide whether to participate, members of the research team explain details of the study. The research team provides an informed consent document, which includes such details about the study as its purpose, duration, required procedures, and who to contact for various purposes. The informed consent document also explains risks and potential benefits.
If the participant decides to enroll in the trial, the informed consent document will be signed. Informed consent is not a contract. Volunteers are free to withdraw from the study at any time.
IRB review
Most, but not all, clinical trials in the United States are approved and monitored by an Institutional Review Board (IRB) in order to ensure that the risks are minimal and are worth any potential benefits. An IRB is an independent committee that consists of physicians, statisticians, and members of the community who ensure that clinical trials are ethical and that the rights of participants are protected. Potential research participants should ask the sponsor or research coordinator whether the research they are considering participating in was reviewed by an IRB.
Further reading
For more information about research protections, see:
After a clinical trial is completed, the researchers carefully examine information collected during the study before making decisions about the meaning of the findings and about further testing. After a phase I or II trial, the researchers decide whether to move on to the next phase or to stop testing the agent or intervention because it was unsafe or ineffective. When a phase III trial is completed, the researchers examine the data and decide whether the results have medical importance.
Results from clinical trials are often published in peer-reviewed scientific journals. Peer review is a process by which experts review the report before it is published to ensure that the analysis and conclusions are sound. If the results are particularly important, they may be featured in news media and discussed at scientific meetings and by patient advocacy groups before they are published. Once a new approach has been proven safe and effective in a clinical trial, it may become the standard of medical practice.
Ask the research team members if the study results have been or will be published. Published study results are also available by searching for the study's official name or Protocol ID number in the National Library of Medicine's PubMed database.
How does the outcome of clinical research make a difference?
monkeybusinessimages/iStock
Only through clinical research can we gain insights and answers about the safety and effectiveness of drugs and therapies. Groundbreaking scientific advances in the present and the past were possible only because of participation of volunteers, both healthy and those diagnosed with an illness, in clinical research. Clinical research requires complex and rigorous testing in collaboration with communities that are affected by the disease. As clinical research opens new doors to finding ways to diagnose, prevent, treat, or cure disease and disability, clinical trial participation of volunteers is essential to help us find the answers.
ClinicalTrials.gov [ Tips for finding trials on ClinicalTrials.gov ] This is a searchable registry and results database of federally and privately supported clinical trials conducted in the United States and around the world. ClinicalTrials.gov gives you information about a trial's purpose, who may participate, locations, and phone numbers for more details. This information should be used in conjunction with advice from health care professionals.
At the NIH Clinical Center in Bethesda, Maryland
NIH Clinical Center
Search NIH Clinical Research Studies The NIH maintains an online database of clinical research studies taking place at its Clinical Center, which is located on the NIH campus in Bethesda, Maryland. Studies are conducted by most of the institutes and centers across the NIH. The Clinical Center hosts a wide range of studies from rare diseases to chronic health conditions, as well as studies for healthy volunteers. Visitors can search by diagnosis, sign, symptom or other key words.
Join a National Registry of Research Volunteers
ResearchMatch
ResearchMatch(link is external) This is an NIH-funded initiative to connect 1) people who are trying to find research studies, and 2) researchers seeking people to participate in their studies. It is a free, secure registry to make it easier for the public to volunteer and to become involved in clinical research studies that contribute to improved health in the future.
RSVP HERE https://www.eventbrite.com/e/patient-focused-drug-development-meeting-acute-porphyrias-registration-31068098519
Wednesday - January 18, 2017 @ 10:00:15
We at the APF are getting very excited to see you all in Washington, D.C. in less than TWO MONTHS! Many of you have already RSVPed directly to the APF thank you! In an effort to stay organized and efficient, we have created an event webpage for our FDA meeting. Even if you have already RSVPed to us, please sign up through this website, too. For those of you that have not RSVPed, in addition to signing up on this website, please still let Jessica at the APF know that you are planning to attend. She can be reached by email atJessica@porphyriafoundation.org or by phone at the APF office at 866-APF-3635 or 713-266-9617. We will keep you all up to date as the event approaches.
Please do not hesitate to contact us if you have ANY questions surrounding this important event.
Porphyrias are rare disorders that affect mainly the skin or nervous system and may cause abdominal pain. These disorders are usually inherited, meaning they are caused by abnormalities in genes passed from parents to children. When a person has a porphyria, cells fail to change body chemicals called porphyrins and porphyrin precursors into heme, the substance that gives blood its red color. The body makes heme mainly in the bone marrow and liver. Bone marrow is the soft, spongelike tissue inside the bones; it makes stem cells that develop into one of the three types of blood cellsred blood cells, white blood cells, and platelets.
The process of making heme is called the heme biosynthetic pathway. One of eight enzymes controls each step of the process. The body has a problem making heme if any one of the enzymes is at a low level, also called a deficiency. Porphyrins and porphyrin precursors of heme then build up in the body and cause illness.
What is heme and what does it do?
Heme is a red pigment composed of iron linked to a chemical called protoporphyrin. Heme has important functions in the body. The largest amounts of heme are in the form of hemoglobin, found in red blood cells and bone marrow. Hemoglobin carries oxygen from the lungs to all parts of the body. In the liver, heme is a component of proteins that break down hormones, medications, and other chemicals and keep liver cells functioning normally. Heme is an important part of nearly every cell in the body.
What are the types of porphyria?
Each of the eight types of porphyria corresponds to low levels of a specific enzyme in the heme biosynthetic pathway. Experts often classify porphyrias as acute or cutaneous based on the symptoms a person experiences:
Acute porphyrias affect the nervous system. They occur rapidly and last only a short time.
Cutaneous porphyrias affect the skin.
Two types of acute porphyrias, hereditary coproporphyria and variegate porphyria, can also have cutaneous symptoms.
Experts also classify porphyrias as erythropoietic or hepatic:
In erythropoietic porphyrias, the body overproduces porphyrins, mainly in the bone marrow.
In hepatic porphyrias, the body overproduces porphyrins and porphyrin precursors, mainly in the liver.
Table 1 lists each type of porphyria, the deficient enzyme responsible for the disorder, and the main location of porphyrin buildup.
The exact rates of porphyria are unknown and vary around the world. For example, porphyria cutanea tarda is most common in the United States, and variegate porphyria is most common in South America.1
What causes porphyria?
Most porphyrias are inherited disorders. Scientists have identified genes for all eight enzymes in the heme biosynthetic pathway. Most porphyrias result from inheriting an abnormal gene, also called a gene mutation, from one parent. Some porphyrias, such as congenital erythropoietic porphyria, hepatoerythropoietic porphyria, and erythropoietic protoporphyria, occur when a person inherits two abnormal genes, one from each parent. The likeliness of a person passing the abnormal gene or genes to the next generation depends on the type of porphyria.
Porphyria cutanea tarda is usually an acquired disorder, meaning factors other than genes cause the enzyme deficiency. This type of porphyria can be triggered by
too much iron
use of alcohol or estrogen
smoking
chronic hepatitis Ca long-lasting liver disease that causes inflammation, or swelling, of the liver
HIVthe virus that causes AIDS
abnormal genes associated with hemochromatosisthe most common form of iron overload disease, which causes the body to absorb too much iron
For all types of porphyria, symptoms can be triggered by
use of alcohol
smoking
use of certain medications or hormones
exposure to sunlight
stress
dieting and fasting
What are the symptoms of porphyria?
Some people with porphyria-causing gene mutations have latent porphyria, meaning they have no symptoms of the disorder. Symptoms of cutaneous porphyrias include
oversensitivity to sunlight
blisters on exposed areas of the skin
itching and swelling on exposed areas of the skin
Symptoms of acute porphyrias include
pain in the abdomenthe area between the chest and hips
pain in the chest, limbs, or back
nausea and vomiting
constipationa condition in which an adult has fewer than three bowel movements a week or a child has fewer than two bowel movements a week, depending on the person
urinary retentionthe inability to empty the bladder completely
confusion
hallucinations
seizures and muscle weakness
Symptoms of acute porphyrias can develop over hours or days and last for days or weeks. These symptoms can come and go over time, while symptoms of cutaneous porphyrias tend to be more continuous. Porphyria symptoms can vary widely in severity.
How is porphyria diagnosed?
A health care provider diagnoses porphyria with blood, urine, and stool tests. These tests take place at a health care providers office or a commercial facility. A blood test involves drawing blood and sending the sample to a lab for analysis. For urine and stool tests, the patient collects a sample of urine or stool in a special container. A health care provider tests the samples in the office or sends them to a lab for analysis. High levels of porphyrins or porphyrin precursors in blood, urine, or stool indicate porphyria. A health care provider may also recommend DNA testing of a blood sample to look for known gene mutations that cause porphyrias.
How is porphyria treated?
Treatment for porphyria depends on the type of porphyria the person has and the severity of the symptoms.
Acute Porphyrias
A health care provider treats acute porphyrias with heme or glucose loading to decrease the livers production of porphyrins and porphyrin precursors. A patient receives heme intravenously once a day for 4 days. Glucose loading involves giving a patient a glucose solution by mouth or intravenously. Heme is usually more effective and is the treatment of choice unless symptoms are mild. In rare instances, if symptoms are severe, a health care provider will recommend liver transplantation to treat acute porphyria. In liver transplantation, a surgeon removes a diseased or an injured liver and replaces it with a healthy, whole liver or a segment of a liver from another person, called a donor. A patient has liver transplantation surgery in a hospital under general anesthesia. Liver transplantation can cure liver failure. More information is provided in the NIDDK health topic, Liver Transplantation.
Cutaneous Porphyrias
The most important step a person can take to treat a cutaneous porphyria is to avoid sunlight as much as possible. Other cutaneous porphyrias are treated as follows:
Porphyria cutanea tarda. A health care provider treats porphyria cutanea tarda by removing factors that tend to activate the disease and by performing repeated therapeutic phlebotomies to reduce iron in the liver. Therapeutic phlebotomy is the removal of about a pint of blood from a vein in the arm. A technician performs the procedure at a blood donation center, such as a hospital, clinic, or bloodmobile. A patient does not require anesthesia. Another treatment approach is low-dose hydroxychloroquine tablets to reduce porphyrins in the liver.
Erythropoietic protoporphyria. People with erythropoietic protoporphyria may be given beta-carotene or cysteine to improve sunlight tolerance, though these medications do not lower porphyrin levels. Experts recommend hepatitis A and B vaccines and avoiding alcohol to prevent protoporphyric liver failure. A health care provider may use liver transplantation or a combination of medications to treat people who develop liver failure. Unfortunately, liver transplantation does not correct the primary defect, which is the continuous overproduction of protoporphyria by bone marrow. Successful bone marrow transplantations may successfully cure erythropoietic protoporphyria. A health care provider only considers bone marrow transplantation if the disease is severe and leading to secondary liver disease.
Congenital erythropoietic porphyria and hepatoerythropoietic porphyria. People with congenital erythropoietic porphyria or hepatoerythropoietic porphyria may need surgery to remove the spleen or blood transfusions to treat anemia. A surgeon removes the spleen in a hospital, and a patient receives general anesthesia. With a blood transfusion, a patient receives blood through an intravenous (IV) line inserted into a vein. A technician performs the procedure at a blood donation center, and a patient does not need anesthesia.
Secondary Porphyrinurias
Conditions called secondary porphyrinurias, such as disorders of the liver and bone marrow, as well as a number of drugs, chemicals, and toxins are often mistaken for porphyria because they lead to mild or moderate increases in porphyrin levels in the urine. Only highnot mild or moderatelevels of porphyrin or porphyrin precursors lead to a diagnosis of porphyria.
Eating, Diet, and Nutrition
People with an acute porphyria should eat a diet with an average-to-high level of carbohydrates. The recommended dietary allowance for carbohydrates is 130 g per day for adults and children 1 year of age or older; pregnant and breastfeeding women need higher intakes.2 People should avoid limiting intake of carbohydrates and calories, even for short periods of time, as this type of dieting or fasting can trigger symptoms. People with an acute porphyria who want to lose weight should talk with their health care providers about diets they can follow to lose weight gradually.
People undergoing therapeutic phlebotomies should drink plenty of milk, water, or juice before and after each procedure.
A health care provider may recommend vitamin and mineral supplements for people with a cutaneous porphyria.
Points to Remember
Porphyrias are rare disorders that affect mainly the skin or nervous system and may cause abdominal pain.
Each of the eight types of porphyria corresponds to low levels of a specific enzyme in the heme biosynthetic pathway.
The exact rates of porphyria are unknown and vary around the world. For example, porphyria cutanea tarda is most common in the United States, and variegate porphyria is most common in South America.
Most porphyrias result from inheriting an abnormal gene, also called a gene mutation, from one parent.
Porphyria cutanea tarda is usually an acquired disorder, meaning factors other than genes cause the enzyme deficiency.
Symptoms of cutaneous porphyrias include
oversensitivity to sunlight
blisters on exposed areas of the skin
itching and swelling on exposed areas of the skin
Symptoms of acute porphyrias include
pain in the abdomen
pain in the chest, limbs, or back
nausea and vomiting
constipation
urinary retention
confusion
hallucinations
seizures and muscle weakness
A health care provider diagnoses porphyria with blood, urine, and stool tests.
Treatment for porphyria depends on the type of porphyria the person has and the severity of the symptoms.
References
Clinical Trials
The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and other components of the National Institutes of Health (NIH) conduct and support research into many diseases and conditions.
What are clinical trials, and are they right for you?
Clinical trials are part of clinical research and at the heart of all medical advances. Clinical trials look at new ways to prevent, detect, or treat disease. Researchers also use clinical trials to look at other aspects of care, such as improving the quality of life for people with chronic illnesses. Find out if clinical trials are right for you .
What clinical trials are open?
Clinical trials that are currently open and are recruiting can be viewed at www.ClinicalTrials.gov.
Porphyria is not a single disease but a group of at least eight disorders that differ considerably from each other. A common feature in all porphyrias is the accumulation in the body of porphyrins or porphyrin precursors. Although these are normal body chemicals, they normally do not accumulate. Precisely which of these chemicals builds up depends on the type of porphyria.
The terms porphyrin and porphyria are derived from the Greek word porphyrus, meaning purple. Urine from some porphyria patients may be reddish in color due to the presence of excess porphyrins and related substances in the urine, and the urine may darken after exposure to light.
The symptoms and treatment vary significantly from one type of Porphyria to the next.
Porphyria symptoms arise mostly from effects on either the nervous system or the skin. Effects on the nervous system occur in the acute porphyrias (AIP, ADP, HCP and VP). Proper diagnosis is often delayed because the symptoms are nonspecific. Skin manifestations can include burning, blistering and scarring of sun-exposed areas.
The porphyrias are rare diseases. Taken together, all forms of porphyria afflict fewer than 200,000 people in the United States. Based on European studies, the prevalence of the most common porphyria, porphyria cutanea tarda (PCT), is 1 in 10,000, the most common acute porphyria, acute intermittent porphyria (AlP), is about 1 in 20,000, and the most common erythropoietic porphyria, erythropoietic protoporphyria (EPP), is estimated at 1 in 50,000 to 75,000. Congenital erythropoietic porphyria (CEP) is extremely rare with prevalence estimates of 1 in 1,000,000 or less. Only 6 cases of ALAD-deficiency porphyria (ADP) are documented.
EPP is the most common porphyria in childhood, and the one associated with the longest delays in diagnosis.
PSA Get Well
Wednesday - January 4, 2017 @ 13:32:47
We know that many this time of year are sick. This can be the worst time for an attack, due to the holidays, travel, illness, winter and stress. Please let us know how we may may be able to help you by providing your Dr with a comprehensive Dr Packet or a patient kit for yourself. Our thoughts and prayers are with you all during such difficult times
Porphyria is not a single disease but group of no
Wednesday - January 4, 2017 @ 21:01:26
Porphyria is not a single disease but group of no less than eight issue that contrast considerably from each other. Our experts have identified a formula which is purely herbal and particularly appropriate for the Porphyria Herbal Treatment. The reasons why choose us for curing Porphyria is because we offer an ultimate Herbal Supplements for this disease.
For just over 40 years I've been using zinc sulphate to deactivate the PBG my AIP produces that poisons my nervous system, about 12-15 mg orally when I have symptoms. It's cheap, effective, available.
What a great suggestion. I want to try this and se
Sunday - January 22, 2017 @ 22:55:52
What a great suggestion. I want to try this and see if it will help. What brand are you using that helps? Also, do you have any idea or theory as to exactly why it helps? Thank you so much.
(I tried to edit this earlier to add an additional question instead it deleted it. Sorry.)
I am so excited to be able to attend a be a voice
Tuesday - April 4, 2017 @ 11:09:13
I am so excited to be able to attend a be a voice for all of us Porphyria Patients. I'm really praying for all of us attending to make such an impact that this drug will be approved and available ASAP. It is a new Much Needed HOPE !!
Excellent resource!
Friday - May 5, 2017 @ 08:06:06
Excellent resource!
The best medicine is Scenesse! Epp Sufferers from
Friday - May 12, 2017 @ 14:45:05
The best medicine is Scenesse! Epp Sufferers from the Netherlands, Austria, Switzerland, Germany, Italy and other countries collective find it LIFE CHANGING. After so many years they finally can lead a normal life.
Hopefully this year approval of Scenesse, a very s
Friday - June 2, 2017 @ 08:52:31
Hopefully this year approval of Scenesse, a very succesfull medicine for EPP patients. Without almost any side effects but lifechanging according to many European patients.
I feel your pain - literally. My kids are now 27 &
Thursday - June 22, 2017 @ 06:24:31
I feel your pain - literally. My kids are now 27 & 25 and we took many vacations. My kids are pretty protective of me now but it was a long journey. I remember well the agony of being an EPP parent in the summer and on vacations. You need a plan that protects you first or all suffer. Trust me on this one. Your spouse/family really needs to step up. 'Mom-in-charge' has to pull WAY back when it is vacation time. I know, too, how hard it is not to fall into 'poor me' when you are inside (as ever) and everyone else is outside playing. Hang on! Wonderful memories are also made around the campfire, rainy days, movie day, and sitting in the lodge playing games. Sending all my love because I know vacations are the worst fun of all. :)
Natural Herbs Clinic is a useful source of
Thursday - June 22, 2017 @ 06:27:09
Natural Herbs Clinic is a useful source of Treatment for Porphyria information, Porphyria Herbal Remedy is one of the best herbal remedies which use for the treatment of Porphyria. It treat the disease with out any side effects. Because it made with herbal ingredients.
Natural Herbs Clinic is a useful source of
Thursday - June 22, 2017 @ 06:27:40
Natural Herbs Clinic is a useful source of Treatment for Porphyria information, Porphyria Herbal Remedy is one of the best herbal remedies which use for the treatment of Porphyria. It treat the disease with out any side effects. Because it made with herbal ingredients.
Natural Herbs Clinic is a useful source of
Thursday - June 22, 2017 @ 06:28:06
Natural Herbs Clinic is a useful source of Treatment for Porphyria information, Porphyria Herbal Remedy is one of the best herbal remedies which use for the treatment of Porphyria. It treat the disease with out any side effects. Because it made with herbal ingredients.
Natural Herbs Clinic is a useful source of
Thursday - June 22, 2017 @ 06:33:00
Natural Herbs Clinic is a useful source of Treatment for Porphyria information, Porphyria Herbal Remedy is one of the best herbal remedies which use for the treatment of Porphyria. It treat the disease with out any side effects. Because it made with herbal ingredients.
Natural Herbs Clinic is a useful source of
Thursday - June 22, 2017 @ 06:33:21
Natural Herbs Clinic is a useful source of Treatment for Porphyria information, Porphyria Herbal Remedy is one of the best herbal remedies which use for the treatment of Porphyria. It treat the disease with out any side effects. Because it made with herbal ingredients.
Natural Herbs Clinic is a useful source of
Thursday - June 22, 2017 @ 06:35:00
Natural Herbs Clinic is a useful source of Treatment for Porphyria information, Porphyria Herbal Remedy is one of the best herbal remedies which use for the treatment of Porphyria. It treat the disease with out any side effects. Because it made with herbal ingredients.
Natural Herbs Clinic is a useful source of
Thursday - June 22, 2017 @ 06:35:46
Natural Herbs Clinic is a useful source of Treatment for Porphyria information, Porphyria Herbal Remedy is one of the best herbal remedies which use for the treatment of Porphyria. It treat the disease with out any side effects. Because it made with herbal ingredients.
Natural Herbs Clinic is a useful source of
Thursday - June 22, 2017 @ 06:36:06
Natural Herbs Clinic is a useful source of Treatment for Porphyria information, Porphyria Herbal Remedy is one of the best herbal remedies which use for the treatment of Porphyria. It treat the disease with out any side effects. Because it made with herbal ingredients.
Natural Herbs Clinic is a useful source of
Thursday - June 22, 2017 @ 06:36:15
Natural Herbs Clinic is a useful source of Treatment for Porphyria information, Porphyria Herbal Remedy is one of the best herbal remedies which use for the treatment of Porphyria. It treat the disease with out any side effects. Because it made with herbal ingredients.
Natural Herbs Clinic is a useful source of
Thursday - June 22, 2017 @ 06:37:29
Natural Herbs Clinic is a useful source of Treatment for Porphyria information, Porphyria Herbal Remedy is one of the best herbal remedies which use for the treatment of Porphyria. It treat the disease with out any side effects. Because it made with herbal ingredients.
Natural Herbs Clinic is a useful source of
Thursday - June 22, 2017 @ 06:37:33
Natural Herbs Clinic is a useful source of Treatment for Porphyria information, Porphyria Herbal Remedy is one of the best herbal remedies which use for the treatment of Porphyria. It treat the disease with out any side effects. Because it made with herbal ingredients.
Natural Herbs Clinic is a useful source of
Thursday - June 22, 2017 @ 06:39:18
Natural Herbs Clinic is a useful source of Treatment for Porphyria information, Porphyria Herbal Remedy is one of the best herbal remedies which use for the treatment of Porphyria. It treat the disease with out any side effects. Because it made with herbal ingredients.
Natural Herbs Clinic is a useful source of
Thursday - July 13, 2017 @ 06:52:20
Natural Herbs Clinic is a useful source of Treatment for Porphyria information, Porphyria Herbal Remedy is one of the best herbal remedies which use for the treatment of Porphyria. It treat the disease with out any side effects. Because it made with herbal ingredients.
Granuloma annulare is a kind of chronic skin disor
Tuesday - July 25, 2017 @ 03:08:50
Granuloma annulare is a kind of chronic skin disorder characterized by the raised.This great sign shows that your body is healing properly these tips and Granuloma Annulare Natural Treatment you don't have to worry about the unwanted side effects because every one of the ingredients utilized are natural and no potions.
Hi Today I want to show you a really absurd, but
Thursday - July 27, 2017 @ 00:20:20
Hi
Today I want to show you a really absurd, but extremely effective 'Manifestation Formula'
This will FORCE the universe to give you the life of your dreams - money, happiness, success, and more
Whether your wish is for more wealth, success, happiness, better relationships, good health Make sure you download your kit while this is still available:
==> Go here for The Manifestation Breakthrough Kit [YOUR LINK HERE]
I particularly like the mp3 audio file that comes with it, which puts the Law of Attraction on autopilot to manifest prosperity, abundance and happiness.
The life of your dreams is yours!
P.S. I'm not sure how much longer this will be available for free so download your copy as soon as you can:
Wonderful! I've suffered from AIP since age 9 when I was administered sulfa drugs for tonsillitis, triggering months of hell worlds and sensitizing me to numerous other attacks before I was diagnosed at age 26. Diagnosis is the key. Glad you got the care you needed!
And stop eating and drinking foods or beverages in
Wednesday - October 4, 2017 @ 09:08:18
And stop eating and drinking foods or beverages in plastic packaging. Plastic, particularly PET #1 plastic, leaches hormone disrupters into the food and drink, increasing risk of diabetes, obesity, low sperm count, various forms of cancer, and can trigger AIP attacks.
I suspect studies of the US prison population (whe
Wednesday - October 25, 2017 @ 07:14:59
I suspect studies of the US prison population (where the US houses most of its mentally ill) would reveal a large number of undiagnosed cases of AIP. Change their diet, start zinc sulfate therapy, get them into a supportive environment and they could become whole again.
This article seems important but without a one par
Friday - November 3, 2017 @ 23:47:59
This article seems important but without a one paragraph layman's interpretation I just don't know if I should be concerned or if I should just file away for reference.

































































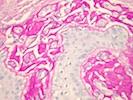
















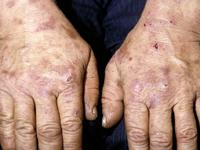
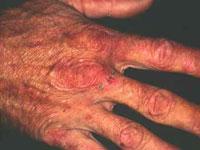

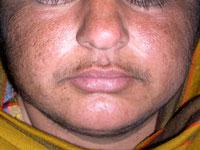


































 = Normal UROS gene
= Normal UROS gene  = Mutated UROS gene
= Mutated UROS gene












































{kind=link}
{kind=link}
{kind=link}
{kind=link}