Welcome to Shadow Jumpers! This is a page for EPP kids and parents by EPP kids and parents. Shadow Jumpers was created to help give kids with Erythropoietic Protoporphyria (EPP) and their families a place to learn about this rare disease, read tips and tricks learned over time and to hear from fellow kids. Through spreading awareness, fellow EPP interviews, tips to protecting yourself outside and some insight for parents, we hope all families living with EPP will look at this condition as a challenge they can overcome. Be sure to check out all the Shadow Jumper resources! Wed love to hear from you. Reach out to us anytime on shadowjumpers@porphyriafoundation.org!!
A main goal of Shadow Jumpers is to help kids living with Erythropoietic Protoporphyria do things they have always wanted to do but have not been able because of the sun. There are only several hundred diagnosed cases of this ultra-rare genetic disorder in the United States so we need to learn from and support one another! Pain from the sun combined with the mental discomfort kids can feel from having to cover up may lead kids to avoid certain activities, relationships and paths in life.
Shadow Jumpers wants to show (and remind) kids that they can still follow their dreams! Whether thats trying to play a sport outdoors, going on a family vacation, camping or whatever comes to mind, Shadow Jumpers wants to help. Click around to learn about some awesome EPP kids, how we are trying to do our part and how you can too!
Here are ways to connect with Shadow Jumpers!
Importance of Medical IDs
Thursday - December 27, 2018 @ 06:31:52
Importance of Medical IDs
Your medical ID provides for a quick recognition of your medical conditions, allergies, medications, or treatment wishes; this leads to faster and more effective medical treatment.
Medical ID bracelets reduce treatment errors which may result from not having a patients health record during an emergency situation or upon hospital admission.
A medical ID speaks for you in the event of an emergency if you become unresponsive
First responders and medical personnel are trained to first look for medical identification jewelry in an emergency. Medical IDs will immediately alert emergency medical professionals to your critical health and personal information.
There are unlimited reasons for you and your loved ones to wear a medical ID when living with common or unusual medical ailments. A medical ID will save your life and the lives of those you love.
Caring for a loved one suffering from a medical condition can strain even the most resilient people. Caregiving can have many rewards while also taking a substantial toll on both the caregiver and their loved ones.
Common Signs of Caregiver Stress
Because caregivers are most commonly altruistic in nature, you may be completely focused on the health and welfare of your loved one before realizing your own overall well-being is suffering. When taking upon the role as a family caregiver, try to be cognizant of early warning signs of caregiver stress such as:
Feeling overwhelmed or constantly worried
Feeling tired most of the time
Sleeping too much or too little
Gaining or losing a lot of weight
Becoming easily irritated or angry
Losing interest in activities you used to enjoy
Feeling sad
Having frequent headaches, bodily pain or other physical problems
Abusing alcohol or drugs, including prescription medications
Excess stress over an extended period of time can do more than cause increased depression and anxiety. Lack of sleep, eating a poor diet, and not getting enough physical activity can increase your chance of developing serious medical problems including heart disease and diabetes.
Strategies for Managing Caregiver Stress
Ask for help- dont automatically assume you have to take on everything. Make a list of priorities and reach out to friends, other family members and even distant relatives.
Take daily breaks- You deserve it. Enjoy some you time throughout the day whether its going to the gym, reading a book, or engaging in another hobby.
Just say no- Accept the fact that you simply cant do everything. Resist the urge to take on more activities, projects or financial obligations that you feel you can handle. Be honest when feeling you are being stretched too thin.
Get organized- try to keep your responsibilities prioritized. Dont stress too much if you cant manage everything and just take care of the most important things one at a time.
Stay connected with friends and family- try to keep yourself from being isolated from family and friends. It can be easy to completely absorb yourself into your role as caregiver increasing your stress level. Make time for social gatherings- dinner with friends, attending family events and holiday celebrations, etc.
Keep a positive attitude- When caring for another living with a chronic medical condition, it can be easy to fall into a negative mindset. Instead of dwelling on what you cant control, give yourself praise for how much you are impacting the life of your loved one in need.
Set personal health goals- establishing a normal sleep pattern, regularly finding time to be physically active and fueling your body with a balanced diet are great ways to battle stress while maintaining your own emotional and physical health.
Join a caregiver support group- others in a support group can understand your trials and frustrations while offering alternative solutions. A support group can present opportunities to interact with others in order to avoid isolation; while giving you the chance to create meaningful relationships.
https://www.caring.com/support-groups offers many featured online groups for caregivers, offering you an outlet to share, vent, laugh, and feel less alone.
The porphyrias are inherited genetic conditions, which means that people with a porphyria have changes to certain genes that affect their bodys ability to regulate itself. When genes are copied, either to make new cells or to make a child, sometimes the body makes an imperfect copy. There can be little changes in the genes, called mutations, which can occur randomly. Sometimes these changes do not make any difference in how well the gene works, but other times they can keep the gene from working properly (referred to as mutations) and are disease causing.
In the porphyrias, these mutations are in the genes involved in a certain chemical pathway, called the heme biosynthetic pathway. Heme is a compound that the body needs to make hemoglobin and there are several steps to make this compound in the body. Each type of porphyria is caused by a defect in a specific enzyme in the heme biosynthetic pathway. Without these enzymes working properly, the body is not able to finish making heme and it causes a buildup of other compounds, called porphyrins. It is the buildup of different types of porphyrins that causes the different types of porphyria.
Classification of the Porphyrias
Most commonly the porphyrias are divided into the acute and cutaneous porphyrias, depending on the primary symptoms. The acute porphyrias [acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), variegate porphyria (VP), and ALA-dehydratase deficiency porphyria (ALD)] present with sudden attacks of severe stomach pain that last for several days; VP and HCP may also have skin symptoms of blistering after sun exposure. The cutaneous porphyrias present with blistering and scarring of the skin, pain, and/or redness and swelling in sun-exposed areas. The porphyrias may also be classified as hepatic or erythropoietic, depending on the organ where the porphyrins accumulate, the liver for the hepatic porphyrias [AIP, HCP, VP, porphyria cutanea tarda (PCT), and hepatoerythropoietic porphyria (HEP)] or the bone marrow for the erythropoietic porphyrias [congenital erythropoietic porphyria (CEP), erythropoietic protoporphyria (EPP), and X-linked protoporphyria (XLP)].
The Acute Porphyrias
There are four types of acute porphyrias; acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), variegate porphyria (VP), and δ-aminolevulinic acid dehydratase porphyria (ADP), and they have similar symptoms. These are genetic disorders that are very rare and may be difficult to diagnose for this reason. It is estimated that about 1 in 10,000 Europeans or people of European ancestry have a mutation in one of the genes that cause AIP, VP or HCP. These mutations have been found in all races and many other ethnicities in addition to Europeans.
Approximately 80-90% of individuals who carry a gene mutation for acute intermittent porphyria, variegate porphyria, and hereditary coproporphyria, remain asymptomatic, and others may have only one or a few acute attacks throughout life. The most frequent symptom is severe abdominal pain and is often accompanied by nausea, vomiting, and constipation. Other symptoms may include heart palpitations, seizures, and hallucinations. People with VP and HCP may also have skin symptoms of blistering after sun exposure.
The Cutaneous Porphyrias
All but one of the cutaneous porphyrias cause skin blistering and fragility on sun-exposed areas of the body, most commonly the backs of the hands, forearms, face, ears and neck. The cutaneous porphyrias are porphyria cutanea tarda (PCT), hepatoerythropoietic porphyria (HEP), congenital erythropoietic porphyria (CEP), erythropoietic protoporphyria (EPP), and X-linked protoporphyria (XLP).
CEP and HEP occur in childhood with severe blistering skin lesions. PCT occurs in adulthood generally and less severe blistering skin lesions after sun exposure. Erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) have the same symptoms of painful, but nonblistering, reactions to sunlight. There can also be swelling and redness of the sun exposed areas of the skin with EPP and XLP.
Inheritance of the Porphyrias
Each type of porphyria is caused by a mutation, or change, in the gene coding for a specific enzyme in the heme pathway. PCT is unique as it is the only porphyria where most patients do not have mutations in a gene, but instead have acquired, or sporadic, PCT.
Types of porphyria, their patterns of inheritance, and the enzyme that is deficient in each.
The inherited porphyrias are either autosomal dominant (inherited from one parent), autosomal recessive (inherited from both parents), or X-linked (the gene is located on the X-chromosome). "Autosomal" genes always occur in pairs, with one coming from each parent. Individuals with an autosomal dominant form of porphyria have one mutated gene paired with a normal gene, and there is a 50% chance with each pregnancy that the mutated gene will be passed to a child.
Individuals with an autosomal recessive type of porphyria have mutations on both copies of a specific gene, one passed to them from each of their parents. Each of their children will inherit one mutated gene for that porphyria, and the child will be a carrier but will not have symptoms.
In X-linked disorders, the gene is located on one of the sex chromosomes, called the X-chromosome. Females have two X-chromosomes, and males have one X-chromosome and one Y-chromosome. Both males and females will likely have symptoms from a mutated gene on the X-chromosome, but females, with a normal gene on the other X-chromosome, usually are less severely affected than males. The risk for children depends on the gender of the affected parent. A female with an X-linked gene mutation will have a 50% risk of passing that mutation to any of her children with each pregnancy. However, a male will pass the mutation to all of his daughters but none of his sons.
Diagnosis of the Porphyrias
There are many laboratory tests available for the porphyrias, and the right tests to order depend on the type of porphyria the doctor suspects. When abdominal and neurological symptoms suggest an acute porphyria, the best screening tests are urinary aminolevulinic acid (ALA) and porphobilinogen (PBG). When there are cutaneous symptoms that suggest porphyria, the best screening test is a plasma porphyrin assay. If one of these screening tests is abnormal, more extensive testing, including urinary, fecal, and red blood cell porphyrins, are often indicated.
DNA testing to identify the specific mutation in an individuals porphyria-causing gene is also recommended. Before requesting DNA testing, it is helpful that patients have biochemical testing. However, many patients have not had an acute attack or are not symptomatic at present, so biochemical testing may be inconclusive.
In contrast, DNA testing is the most accurate and reliable method for determining if a person has a specific porphyria and is considered the "gold standard" for the diagnosis of genetic disorders. If a mutation (or change) in the DNA sequence is found in a specific Porphyria-causing gene, the diagnosis of that Porphyria is confirmed. DNA analysis will detect more than 97% of disease-causing mutations. DNA testing can be performed whether the patient is symptomatic or not. Once a mutation has been identified, DNA analysis can then be performed on other family members to determine if they have inherited that Porphyria, thus allowing identification of individuals who can be counseled about appropriate management in order to avoid or minimize disease complications.
Acute Intermittent Porphyria (AIP) is an inherited genetic condition. The genetic mutations that cause AIP are in the HMBS gene. They result in the genes to produce too little of the enzyme hydroxymethylbilane synthase (also called porphobilinogen deaminase). Without enough of this enzyme, the body is not able to finish converting porphobilinogen into heme chains, causing them to build up to much higher levels than usual. This buildup can cause the pain attacks of AIP, but about 80-90% of patients with AIP mutations will not develop symptoms.
When patients do suffer an attack, they will usually experience severe abdominal pain. This is often extremely painful and patients may need to go to the hospital for help. Because these attacks will often not involve any visible symptoms, the ER staff may not know to treat patients for porphyria if they are unaware of the diagnosis. It is very important for patients to speak with their local hospital and health care provider to make sure a plan is in place before an attack so they are able to receive care as quickly as possible.
Patients can also experience numbness, weakness, nausea, constipation, confusion, restlessness, hallucination, seizures, and difficulty with urination during acute attacks. These symptoms can be very severe and hard to treat if a doctor does not know to suspect AIP. It is important for family members of AIP patients to get tested even if they have never had an attack before. If they do have a mutation in one of their copies of the HMBS gene, knowing this will allow their doctor to give them the appropriate care if any symptoms arise.
Patients with AIP also have a slightly increased risk than the general population of developing liver cancer, called hepatocellular carcinoma.
How is Acute Intermittent Porphyria diagnosed?
There are two types of testing; biochemical, meaning looking for biomarkers in the blood or urine, and genetic, meaning looking at the gene we know causes the disease directly from a blood sample.
Biochemical: To diagnose AIP the most important test is to measure the level of porphobilinogen (PBG) in the urine. This test is often combined with measuring the total amount of porphyrins in the urine and another biomarker called aminolevulinic acid (ALA). The level of PBG in the body can vary so the best time to take samples is during an acute attack (e.g. when someone is having abdominal pain, etc). In people with AIP the level of PBG is very high.
Genetic: A blood sample is used to look at a persons genes and by doing this it is possible to see if their genes have mutations that can cause disease. The gene that causes AIP is called HMBS. Genetic testing is recommended for patients even if they have very high PBG levels.
If a patient has a mutation, their immediate family members should be tested for that same mutation as well. This includes their parents, their siblings, and any children they may have. This will allow all family members to receive appropriate care and counseling even though 80-90% of people with a mutation will not develop symptoms of AIP.
What are treatments for Acute Intermittent Porphyria?
AIP attacks can be triggered by a number of factors. One known trigger is progesterone, a hormone which naturally increases in women during their menstrual cycle. Female AIP patients are more likely to have attacks in the second half of their menstrual cycle, when their uterine lining is thickening but before it begins to shed (when they begin bleeding). Dieting can also be a trigger, so patients should avoid fasting and dieting. Patients with AIP should eat a balanced diet. Drugs can be another trigger, especially barbiturates, sulfonamide antibiotics, anti-seizure drugs, and oral contraceptives (progesterone in particular). There is an online drug database to check which medications may be unsafe for people with AIP. The American Porphyria Foundation offers a mobile phone app that pulls up this information online (porphyriadrugs.com).
During an attack, patients may often need to be hospitalized. This will allow them to receive medications to handle their pain and IV fluids if they are unable to stop vomiting or are too nauseous to eat. If the attack was triggered by using drugs for a long time, the muscles which control breathing may be weak and the patient may need respiratory support.
Patients can receive heme therapy through an IV. Panhematin is an FDA approved medication which can help decrease the severity and length of the attack, and is more effective the earlier they receive it.
Attacks can be prevented in many cases by avoiding harmful drugs and fasting or dieting. Wearing a Medic Alert bracelet is recommended for patients who have had attacks. Very frequent premenstrual attacks can be prevented by a gonadotropin-releasing hormone (GnRH) analogue administered with expert guidance. In some cases, frequent attacks can be prevented by regularly scheduled infusions of hemin.
Individuals with AIP who are prone to attacks should eat a normal balanced diet and should not fast or diet, even for short periods of time. If weight loss is desired, it is advisable to consult a physician and a dietitian to have them an individualized diet plan created.
How is AIP Inherited?
AIP is an autosomal dominant condition. Autosomal means that the defect is not on the chromosomes that determine sex, and dominant means that you only need to inherit one mutated gene to manifest the disease. The gene that causes AIP is called HMBS.
Genes are inherited randomly, so a parent has an equal chance of passing on either of their two copies of each gene. Since most AIP patients have one mutated copy and one normal copy, this means that each of their children will have a 50% chance of inheriting the mutated copy and 50% chance of inheriting the working copy.
Who Make Up Our Scientific Advisory Board
Friday - December 21, 2018 @ 06:30:06
SCIENTIFIC ADVISORY BOARD
Our Scientific Advisory Board is made up of the world's foremost experts in Porphyria management, diagnosis, and research. They have written or approved the medical information on this website. Many of the American Porphyria Foundation's SAB members have over 40 years experience working on Porphyria - conducting cutting-edge research, and writing peer-reviewed article for major medical journals and authoring the chapters on Porphyria in medical school textbooks. Doctors worldwide consult with these Porphyria specialists for help with diagnosis and treatment of their patients.
Karl E. Anderson, MD, Chairman University of Texas Medical Branch
Manisha Balwani, MD Mount Sinai School of Medicine
D. Montgomery Bissell, MD University of California
Joseph R. Bloomer, MD University of Alabama
Herbert L. Bonkovsky, MD Wake Forest Baptist Medical Center Winston-Salem, North Carolina
Sylvia S. Bottomley, MD University of Oklahoma
Robert J. Desnick, PhD, MD Mount Sinai School of Medicine
Micheline M. Mathews-Roth, MD Harvard University School of Medicine
John Phillips, PhD University of Utah
Claus A. Pierach, MD University of Minnesota
Neville Pimstone, MD, PhD University of California
Maureen B. Poh-Fitzpatrick, MD University of Tennessee
Steven Shedlofsky, MD University of Kentucky
Samuel M. Silver, MD, PhD University of Michigan Medical School
Ashwani Singal, MD University of Alabama, Birmingham
Manish Thapar, MD Sidney Kimmel Medical College of Thomas Jefferson University
Peter V. Tishler, MD Harvard University School of Medicine
Bruce Trippe, MD Montgomery, AL
Bruce Wang, MD University of California, San Francisco
Protect the Future
Simon Beaven, MD, PhD University of California, Los Angeles
Brendan Chen, MD Mount Sinai School of Medicine
Maria A. de Lima, MD Rio de Janerio, Brazil
Angelika Erwin, MD Cleveland, Ohio
Bradley Freilich, MD Kansas City, Missouri
Eric Gou, MD University of Texas Medical Branch
Jennifer Guy, MD University of California, San Francisco
Sajid Jilil, MD Fargo, North Dakota
Sioban Keel Seattle Cancer Care Alliance, Univ. WA
Cynthia Levy, MD University of Miami
Charles Marques Lourenço, MD University of São Paulo, Brazil
Brendan McGuire, MD University of Alabama-Birmingham
Sahil Mittal, MD Houston, Texas
Akshata Moghe, MD, PhD University of Pittsburgh
John G Quigley University of Illinois at Chicago
Sean Rudnick, MD Wake Forest Medical Center
Behnam Saber, MD Mount Sinai School of Medicine
Jonathan Ungar, MD Mount Sinai School of Medicine
Happy Winter Season!
Wednesday - December 19, 2018 @ 17:19:53
Happy Winter Season!
Porphyria Post
Friday - December 14, 2018 @ 06:30:00
Porphyria Post
Happy Holidays from the APF!
The American Porphyria Foundation will be closed for the Christmas holiday beginning on Monday, December 24, 2018 and will re-open for normal business hours on Wednesday, January 2, 2019.
For emergency medical attention, please go to your nearest emergency room and contact your primary care physician. For all other issues, please direct your needs to the APF office staff via email. Your messages will be answered accordingly.
Desiree Lyon - lyonapf@aol.com
Kristen Wheeden - kristen@porphyriafoundation.org
We hope that you enjoy this Christmas and New Years holiday with your loved ones.
Reminders!
EPP Clinical Trial Participation - We need YOU!
Please note: CANADIANS ARE NOW ELIGIBLE TO PARTICIPATE!
A clinical trial with an oral investigational drug intended to reduce phototoxicity in people living with EPP is underway.
Please note that a Phase 3 study is very unlikely if Phase 2 is not completed. To date, this drug has proven nontoxic and there have been essentially no dropouts in the Phase 2 study to date.
This is an oral drug which makes administration simple.
This study in adults will pave the way for a near-future pediatric trial. EPP is a rare disease and patients must participate in order to get the drug approved.
To date there is no public information on the availability of Scenesse, and if approved, two drugs on the market may lower their cost.
We will connect you with a Research Coordinator that will answer all your questions and concerns. We will work to make this process as easy as possible for you.
Eligibility Criteria:
â?¢ Confirmed diagnosis of EPP
â?¢ Provide written informed consent to participate
â?¢ Be willing and able to travel to all study sites for scheduled visits
â?¢ 18 - 70 years of age at the time of screening.
Sites enrolling:
â?¢ Mount Sinai - New York City
â?¢ Wake Forest Baptist University - Winston Salem, NC
â?¢ University of Miami - Miami, FL
â?¢ University of Texas Medical Branch - Galveston, TX
â?¢ University of Utah - Salt Lake City, UT
â?¢ University of California San Francisco - San Francisco, CA
We need YOU! Please dig deep and consider being part of this changing moment for all with EPP.
Contact us here at the APF office to get in contact with a research coordinator.
301.347.7166 or 1.866.APF.635
porphyrus@porphyriafoundation.com
"RememberResearch is the key to YOUR cure!"
Each Step Toward Finding an Effective Treatment is Important!
Pet Calendars on Sale!
Order your APF 2019 Pet Calendar TODAY! This is a great way to start your year off right and support the APF.
Calendars are $12 including shipping and can be ordered through the APF Store or by emailing autumnlee@porphyriafoundation.org.
The calendar is filled with your fluffy companions, dates, and porphyria facts.
Click on the link below to order today!
https://porphyriafoundation.org/store
Harvoni Study - PCT
Do you have PCT? Are you interested in participating in research? Do you have Hepatitis C? If you answered YES to these questions, this is for you.
We need YOU for a clinical trial!
The purpose of this clinical trial is to assess whether Harvoni alone is an effective therapy in active PCT patients with Chronic Hepatitis C.
Who can participate?
1. Adult patients with PCT who also have Hepatitis C
If you are interested in participating please contact Edrin Williams, Director of Patient Services at the APF office at 301.347.7166 for additional information.
Get Involved
â?¢ Research â?¢
Contact Information
Is your contact information up to date?
If not please give us a call @ 866-APF-3635 or Email to porphyrus@porphyriafoundation.com
American Porphyria Foundation 1.866.APF.3635 porphyriafoundation.org
STAY CONNECTED
"Remember.Research is the Key to Your Cure!"
The Porphyria story of Victor LaFae with HCP
Thursday - December 13, 2018 @ 18:38:10
Porphyria story - HCP - Victor LaFae
Im told that I was a typical happy baby for the first few months of my life. I reached all my milestones on time or even a little bit early. It was not until I was three months old that something seemed amiss. One Saturday afternoon in mid-July 1993 while my father was at work my mother decided to take me and my older sister out to a park and to run some errands. My sister ran around the park having a blast for a couple of hours while my mother sat in on a bench with me partially shaded by a large oak tree. As the sun began to set my sister was called from the playground and my mother began the walk to the grocery store. I was getting fussy, and she assumed that I was tired and ready to eat and sleep.
About halfway to the store, I let out a screeching noise. Thinking I must have been bitten by a spider or stung by a bee my mother stopped to shake out my blanket and check me over for bites. She found nothing to explain my screaming cry. When I could not be comforted my mother grew more and more concerned. That night we ended up sitting in a hospital for several hours. The doctors ran blood tests and found nothing unusual. They told my mother that I was a colicky baby and that I would outgrow it. Not having raised a colicky child before, my parents grudgingly accepted the doctors' diagnosis and took me home.
A few weeks went by and I continued to scream and cry every time my mother tried to take me out of the house. It was the start of August and my older sister was getting sick of being cooped up at home. So, I was taken to the park to let my sister play. My mother decided that perhaps I just needed to cry it out as she sat under the oak tree talking with another mother whose child was off playing with my sister. When the other woman inquired about my cries, my mother told her I was just colicky. That is not a colicky baby! My boy was colicky, the mother declared. As if to prove the woman's point, I began having my first of many seizures.
I only seized for a moment or two, but it was enough to have my mother rush off to the ER. The doctors again did blood work and asked many questions. They found nothing to explain the seizure. They told her that I was colicky and that I was likely just trembling from having exhausted myself crying. Over the next few months, things would spiral out of control with new and more bizarre symptoms popping up every few days. Each symptom dismissed and my mother became known as the over dramatic nervous mother. With each new visit to the ER, the doctors took my mother less seriously than the last.
I became lethargic. My arms and legs that had once danced around lay limp at my side. I stopped trying to nurse and spit up anything my mother managed to get into me. My weight decreased until I was not even on the growth curve. I didnt react to the things I once loved, like peek a boo. I developed tremors and muscle twitches, small blisters, and dark urine. My mother was told again that it was all from being a colicky baby, I was probably dehydrated, and that the blisters were from the laundry soap. This went on for a few years. Slowly some of the symptoms cleared up and so my parents believed that perhaps the doctors had been right.
At around 4 years old, the first signs of my Autism became clear, and I was later diagnosed with Asperger's syndrome. I was minimally communicative which meant I could not explain my own symptoms to the doctors or to my parents. I would only cry when forced outside. I could not explain the burning pain or the itching. My stomach felt like a hot sword was being twisted around in my gut, but I didnt have the words to tell anyone. My heart would beat so hard and fast I was afraid it would burst out of my chest.
I remember so clearly the exams. Every few months, scopes were shoved up one end or down the other as they looked for things like appendicitis and ulcers. My mother was told that with my autism it was hard to really tell why I was having so many meltdowns and that I was likely overstimulated. This is common for autistic children, so she neednt worry. I developed dizziness and began having regular falls. My coordination declined dramatically, I started dropping things and running into walls. This clumsiness was also attributed to my autism and my mother's concerns dismissed.
When I was 10 years old, I had visual hallucinations of red lights sliding around on the walls. I could hear ripping metal like a car being crushed by a compactor and the sound of electric sparks echoing in my head. The pain was back like my insides were being ripped out. The best way I could explain this terrifying and painful experience to the doctors was to say, The demon eyes are hurting my belly and heart. This led to a 72-hour psychiatric hold on the pediatric psych unit. After three days in the unit, I was discharged with a diagnosis of Autism, PTSD, and Somatic Symptom Disorder. Three weeks later in another ER with another blistering rash, the doctors decided I had scarlet fever from an untreated strep infection. There was no strep test done, they just put me on antibiotics. I took the full course of the meds and began having more of the same symptoms only now they were more severe.
This is when it was decided that my parents must be behind it all. They accused my parents of causing my blisters with boiling water and telling me to tell doctors I was sick. They said my mother had Munchausen's By Proxy. With these new abuse allegations, I was placed in foster care where of course my illness continued only now it was being attributed to anxiety and PTSD from the supposed abuse. I have bounced around from one foster home to the next and overlooked for years. This was a very lonely and dark time for me. I felt guilty that my illness was causing my family so much suffering. The pain and guilt lead me to attempt suicide on several occasions.
My suicide attempt led to another psychiatric hospitalization where I met someone with the same symptoms that I had, at this point my communication skills were improving and I was able to sit and speak with the girl. When I told her, what had been happening to me she told me it sounded like her condition but that It couldnt be the same thing because I was too young, and the disease is rare. She had Hereditary Coproporphyria. Even being in the same hospital and treated by the same doctors I was still not diagnosed or even tested for this disease. Hearing how rare the condition was, I dismissed the idea of telling myself the odds were astronomical.
I spent several more years suffering from symptoms no one could explain, and many refused to believe. In 2015, I contracted Pneumonia and an ear infection. One of the medications I was given was Bactrim, which is a medication that is not safe for Porphyria patients. I ended up being admitted to the hospital. For three days no one could figure out what was happening. I was deteriorating before their eyes and they had no answers. During rounds, one of the doctors looking over my medical records made a joke that it looked like I might just be a vampire. I had heard this joke before by doctors and nurses when they read that I could not be exposed to sunlight without blistering and burning.
Suddenly, one of the medical student's eyes grew super wide and he stuttered out one word, Porphyria. The lead doctor ordered a whole bunch of tests and, just in case, started me on a Dextrose drip. The tests were done several times during my stay, often mishandled, but the treatment was slowly working. The doctor informed me that he believed I had a form of Porphyria but could not offer me a definitive diagnosis at the time.
The word Porphyria sounded familiar, but I had long dismissed the idea. I had decided it must be something else and put the memory out of my mind. I didnt really remember the my earlier encounter until later when I was reading about the disease.
For a year and a half, they tested me and each time something else went wrong. It wasnt until December 2016 that they managed to collect, package, and ship the samples correctly. Two weeks later I got the call from Hematologist/ oncologist confirming the diagnosis. I did, in fact, have Hereditary Coproporphyria, a disease that typically does not appear until after puberty or early adulthood. My biochemical testing had revealed elevated ALA, PBG, and Coproporphyrins in my urine. Coproporphyrins were also found in high levels in my stool samples.
I was hospitalized again a couple of months after receiving my diagnosis, this time it was much different. During prior hospitalizations, I had been treated with D10 which slowly helped me recover. However, during this hospitalization, I was given a treatment called Panhematin. When I started the treatment, I was in and out of consciousness, weak, and too lethargic to move. By my third treatment, I was hanging out at the nurse's station, though still in my wheelchair. I was not magically cured, but I was well on my way to recovering.
Though I was relieved to have an answer, I was terrified of what the disease would mean for the rest of my life. I felt lost until my doctor directed me to The American Porphyria Foundation. Through their Facebook support groups I was able to connect with others facing the same challenges and fears.
I receive Panhematin infusions, and I have reconnected with my family. When it is too sunny for me to go outside, I write stories and hang out in the Facebook groups. My life is far from perfect, but I have an answer, I have support, and I have hope that someday I might even have a cure.
Porphyria Post
Wednesday - December 5, 2018 @ 16:50:14
December 12, 2018
Porphyria Post
EPP Clinical Trials: Volunteers Needed!
We are close, but we need YOU!
A clinical trial with an oral investigational drug intended to reduce phototoxicity in people living with EPP is underway.
Please note that a Phase 3 study is very unlikely if Phase 2 is not completed. To date, this drug has proven nontoxic and there have been essentially no dropouts in the Phase 2 study to date.
This is an oral drug which makes administration simple.
This study in adults will pave the way for a near-future pediatric trial. EPP is a rare disease and patients must participate in order to get the drug approved.
To date there is no public information on the availability of Scenesse, and if approved, two drugs on the market may lower their cost.
We will connect you with a Research Coordinator that will answer all your questions and concerns. We will work to make this process as easy as possible for you.
Eligibility Criteria:
·Confirmed diagnosis of EPP
·Provide written informed consent to participate
·Be willing and able to travel to all study sites for scheduled visits
·18 - 70 years of age at the time of screening.
Sites enrolling:
·Mount Sinai - New York City
·Wake Forest Baptist University - Winston Salem, NC
·University of Miami - Miami, FL
·University of Texas Medical Branch - Galveston, TX
·University of Utah - Salt Lake City, UT
·University of California San Francisco - San Francisco, CA
We need YOU! Please dig deep and consider being part of this changing moment for all with EPP.
Contact us here at the APF office to get in contact with a research coordinator.
Do you have PCT? Are you interested in participating in research? Do you have Hepatitis C? If you answered YES to these questions, this is for you.
We need YOU for a clinical trial!
The purpose of this clinical trial is to assess whether Harvoni alone is an effective therapy in active PCT patients with Chronic Hepatitis C.
Who can participate?
1.Adult patients with PCT who also have Hepatitis C
If you are interested in participating please contact Edrin Williams, Director of Patient Services at the APF office at 301.347.7166 for additional information.
Are you interested in participating in this clinical trial?
Do you have AIP, VP or HCP? Are you interested in participating in research? If so, we are recruiting volunteers for the Panhematin Study.
Our researchers need people to volunteer for the Panhematin study. Completing this study will help prevent insurance companies from not paying for this treatment.
The purpose of this study is to determine if Panhematin is safe and effective for prevention of acute attacks of porphyria.
·Can you predict when your next attack will happen?
·Are you currently receiving prophylactic heme treatment?
Please contact Edrin at the APF office (301.347.7166) if you are interested in participating.
Are you interested in participating in this clinical trial?
It is time is to order your 2019 APF Pet Calendar! Calendars will feature all of our contest pet submissions along with important dates and porphyria facts.
Each calendar is $12 for orders shipped in the US. Want to give a gift that gives back? This is the perfect way to support the porphyria community while brightening the year of your friends and family. We can ship it to the home of a loved one with an included gift message. All funds raised support our Protect the Future program and training up the next generation of porphyria experts.
This is a great chance to support the APF and the future of porphyria while enjoying 12-months of our furry, fuzzy, and even feathery friends.
To order call the APF office at 866.APF.3635 or email autumnlee@porphyriafoundation.org or visit the APF store using the link below.
If you have not already done so, please take a moment and bookmark the APF website as one of your favorites! If this is something that you have done in the past, please remove the old bookmark and replace it with the newly updated website address.
Looking for a great gift this winter. Share with a loved one an APF Hat or Tshirt! Extra cold this winter throw on an APF Hoodie or Zipper Hoodie- Looking good & a great price. Check us out! Are you or your child a Shadow Jumper? We got the goods here this season- what are you waiting for come see us. Give yourself a gift by being prepared when you have a flare or an emergency! Stop by and order your comprehensive emergency kit! Cant put a price on this valuable tool. We have Kits in Acute Porphyrias & Cutaneous Types EPP! Check us out.. https://porphyriafoundation.org/store
Thanks for your support and stopping by
The Porphyria Research Consortium: A Partnership Between the American Porphyria Foundation and Physician Researchers
Friday - November 30, 2018 @ 06:30:03
The Porphyria Research Consortium: A Partnership Between the American Porphyria Foundation and Physician Researchers
By: Desiree Lyon Howe, executive director, American Porphyria Foundation
The American Porphyria Foundation (APF) has been instrumental in facilitating porphyria research for over thirty years. The porphyrias are a group of disorders caused by enzyme deficiencies in the heme biosynthetic pathway that affect the Central Nervous System and/or the skin. The collaborative relationship we enjoy with our Scientific Advisory Board is vital to the success of our research projects. This relationship became even more productive when we formed the Porphyria Research Consortium with these porphyria experts. The Consortium is now funded by the National Institutes of Health Rare Disease Clinical Network.
The success of this partnership was evident during the recent Afamelnotide clinical trials. In only one month, we located and registered 90 Erythropoietic Protoporphyria patients for the Phase II trials and 100 more patients for the Phase III trials. This unprecedented accomplishment was achievable because we maintain a well-developed research strategy that includes the Consortium in every step. Their participation, in turn, substantiates our research efforts and gives credibility to the medical information we publish. Such research and educational validation is important to gain the respect of primary care physicians.
Together, we and the Consortium created the National Porphyria Research Registry as a source of research patients and a means to determine incidence. Prior to the initiation of research projects, we prepare patients by educating them on the value and process of clinical trials, introducing them to the researchers, and encouraging them to join the Registry. We reduce patients fears and provide reassurance about the Registry, clinical trials, the expertise of the researchers, and the merit of the research by publishing articles on our website, in our newsletters, and on social media, (e.g., Facebook, Twitter, and blogs). We use these same methods to tout former research patient volunteers, who by sharing their experience, increase the number of future research volunteers.
Researchers in turn assist us by preparing the clinical trials announcements for patients and physicians, which we then distribute via our vast database. Another unique and successful collaboration occurs at our patient education meetings held around the country, during which we locate and register research participants. Three to five experts volunteer for these gatherings to make presentations, answer questions, register patients for their research, and often collect DNA samples.
Also important to our research efforts are the young physicians who participate in the Protect the Future (PTF) program, an APF-supported, mentor project to train the next generation of porphyria experts. The Consortium educates these physicians so they become well versed in both the clinical and basic sciences of the porphyrias and become ready to take on the role of future academic leaders in the field. As part of their training, PTF physicians also conduct mentored research and serve as clinicians at our satellite clinics where patients are given the rare opportunity to meet with a physician with porphyria expertise. In turn, at these clinics, PTF physicians interact with a new pool of research participants.
Our synergistic relationship with our Scientific Advisory Board of experts is key to the success of our research programs.
GET INVOLVED WITH APF
Wednesday - November 28, 2018 @ 10:46:24
GET INVOLVED WITH APF
Become a member of the American Porphyria Foundation today, and join us in our work for your good health!
When you join the APF, you become an integral part of an organization that empowers patients and helps them on the road to accurate diagnosis, proper care, and some day a cure. Adding your voice to ours makes us all stronger as we address our needs to health care providers, local, state and federal agencies and Congress. If you feel you have porphyria but do not have tests indicating you have one of the diseases, you are welcome to join the APF to keep abreast of new discoveries in diagnostics and treatment.
Members of the American Porphyria Foundation receive:
Our quarterly newsletter news about porphyria research and clinical studies, scientific meetings and member get-togethers, and stories about the doctors working in the porphyria field and about members like you;
Attend APF Patient Educational meetings with Experts Nationwide;
Receive weekly E-News in your email;
Free participation in telephone conference calls with top researchers in the porphyria field;
Knowledgethat you are an important part of keeping reliable medical information about porphyria available to those who are newly diagnosed, or at a crossroads in their porphyria treatment, and frightened;
And much more!
The American Porphyria Foundation relies on member support to sustain programs like this website and other literature written or approved by porphyria experts.
U.S. memberships are $35 annually; US membership suggested annual donation is $35. Donate and receive a FREE Porphyria Live DVD, as well as a number of pertinent brochures. International memberships are $45. The APF is a U.S. 501(c)(3) non-profit organization. All donations to the APF are tax deductible.
To join by phone with your VISA or MasterCard, call us toll-free at 1-866-APF-3635 Monday-Friday from 9:00 a.m. to 4:00 p.m. Central Time. To join by mail or fax, please click here to complete and print the donation form.
Unable to pay? Membership is free to all. The fee is only a suggested donation.
American Porphyria Foundation 4915 St. Elmo Avenue, Suite 105 Bethesda, MD 20814
Work with us!
The American Porphyria Foundation welcomes donations of your time and talents too. Would you like to volunteer or raise money for the APF, organize or attend an event, in addition to informing yourself about the porphyrias? If so, welcome! Youll find lots more information in this section of our website on opportunities to get involved in our, and you can always call our office for help: 301-347-7166.
The APF is here to serve, and were proud to have you with us.
THE GLUCOSE EFFECT IN ACUTE PORPHYRIAS
Monday - November 26, 2018 @ 13:47:55
THE GLUCOSE EFFECT IN ACUTE PORPHYRIAS
The disorders Acute Intermittent Porphyria (AIP), Hereditary Coproporphyria (HCP), Variegate Porphyria (VP) and ALAD Porphyria (ADP) are treated initially with the administration of carbohydrate/glucose. This therapy has its basis in the ability of glucose to decrease porphyrin biosynthesis in the liver.
Glucose can diminish excess excretion of heme precursors, which, in turn, can prevent an attack or can hasten recovery from an attack of the acute porphyrias. Therefore, it is suggested that when patients cannot consume carbohydrates due to nausea or vomiting, glucose should be administered intravenously. Some physicians have prepared a standing order for patients who are prone to attacks to help facilitate intravenous glucose in the emergency room. Often this prevents further hospitalization.
Most patients are knowledgeable about the deleterious affect the wrong drug can have on their bodies and are consequently careful about the medication they ingest, but some do not understand the importance of carbohydrates for prevention and treatment of a Porphyria attack.
Because it is a simple therapy, many patients ignore the significance of their carbohydrate (sugar) intake to suppress disease activity. When the Atkins Diet, which was a high protein/low carbohydrate diet, became popular a number of years ago, many Porphyria patients who adhered to this diet became ill. It soon became apparent that their severely reduced daily carbohydrate count exacerbated their Porphyria. In fact, complying with the Atkins plan precipitated attacks in some previously undiagnosed and non-symptomatic individuals.
Support Giving Tuesday
Wednesday - November 28, 2018 @ 19:10:14
Please support the American Porphyria Foundation today! #GivingTuesdayis a global day of giving focused on organizations that have an impact on something you care about. The APF cares about making each day better for individuals and families living with Porphyria.
Every dollar counts as we work to educate physicians, raise awareness and promote research!!
This year Facebook and PayPal are partnering together to match up to a total of $7 million in donations to US nonprofits on #GivingTuesday!
We appreciate all that our members do each day to help us reach our goals - thank you for considering a donation today. Like, Tag and Share!
The APF is proud to support our members that live across the globe. Many countries around the world have developed organizations to support their local communities. We hope that enjoy the same opportunity to communicate with one another, develop friendships, and learn about porphyria.
Here are links to patient advocacy groups worldwide that offer support to individuals impacted by porphyria.
Neuropathic or nerve pain (NP) is a long-term or chronic pain disease that results from nerve damage. It can be caused by different diseases or conditions. Worldwide nerve pain affects as many as 26 million people. Neuropathic or nerve pain may affect larger areas of the body or it can be restricted to a smaller area, in this case it is called localized neuropathic pain (LNP).
WHAT CAUSES NEUROPATHIC PAIN?
Neuropathic or nerve pain may occur in the absence of an obvious visible cause (e.g. an accident, an injury, a chemical burn). There are several external situations that can directly damage nerves and lead to neuropathic pain, such as:
Neuropathic pain is also a common complication of other diseases, including nerve damage after shingles or herpes zoster infection (postherpetic neuralgia or PHN), nerve damage after HIV infection, nerve damage resulting from diabetes mellitus (diabetic polyneuropathy or DPN), multiple sclerosis, or nerve damage in the spine or lower back (low back pain).
WHAT ARE THE TYPICAL NEUROPATHIC PAIN SYMPTOMS?
Neuropathic pain is a chronic pain condition often described by patients with symptoms such as shooting pain, burning pain, or stabbing pain. It can also feel like pins and needles. In addition, neuropathic or nerve pain patients can suffer from symptoms such as allodynia (when a normally unpainful stimulus such as a light touch or clothing becomes painful) and hyperalgesia (when a mild or moderate stimulus causes severe pain).
HOW IS NEUROPATHIC PAIN DIAGNOSED?
Neuropathic pain is often difficult to diagnose. This can lead to patients with neuropathic or nerve pain being insufficiently treated on a trial and error basis that may persist over many months or even years. A correct and early diagnosis is crucial to find the right treatment and to relieve symptoms of neuropathic pain. It is therefore important that patients describe their symptoms in as much detail as possible to their doctor. As part of this process, the doctor will conduct a physical examination and ask about the medical history of the symptoms.
WHAT CAN PATIENTS DO?
There are treatment options available. It is important to get active. Do you have symptoms that you would describe as burning pain, shooting pain, or stabbing pain? Do you have sensitive skin and/or feel a sensation like pins and needles on the skin?
If you have chronic pain and think that it might be neuropathic or nerve pain, please fill out the my painquestionnaire and see your doctor. It is a very useful tool to improve the communication between patients and doctors, as it supports the doctor in making an accurate diagnosis of the cause of chronic pain. It will be most useful for you to accurately describe your chronic pain, where it occurs on your body and if it is triggered by anything in particular. In preparation for a doctors appointment, patients should thoughtfully complete the my painquestionnaire, print out the results, and discuss them with their doctor. You can read more about possible treatment options here.
Porphyria Post
Thursday - November 15, 2018 @ 06:30:11
Porphyria Post
Happy Thanksgiving!
APF Office Closure
We would like to wish you all a very Happy Thanksgiving! For those of you traveling this Holiday, please have a safe and fun trip.
The American Porphyria Foundation will be closed on Thursday, November 22, 2018 and reopen for normal business hours on Monday, November 26, 2018. Please see below for some additional announcements from the APF.
Harvoni Study - PCT
Do you have PCT? Are you interested in participating in research? Do you have Hepatitis C? If you answered YES to these questions, this is for you.
We need YOU for a clinical trial!
The purpose of this clinical trial is to assess whether Harvoni alone is an effective therapy in active PCT patients with Chronic Hepatitis C.
Who can participate?
â?¢ Adult patients with PCT who also have Hepatitis C
If you are interested in participating please contact Edrin Williams, Director of Patient Services at the APF office at 301.347.7166 for additional information.
Clinical Trial for EPP - We NEED you!
The Mitsubishi Clinical Trial is in danger of being cancelled if we do not fully enroll with 102 participants. We must have full participation in order to push this new drug toward approval. We know that a Phase 3 trial is very unlikely if our small EPP community does not come together to fill this trial. Adults who participate will pave the way for a near-future pediatric trial.
Here are the facts. We need 102 total participants for this clinical trial. There are 500+ known patients with EPP in the US, including children and older adults. 1 in 4 patients meet the age requirement. Of those approximately 400 patients, another subset cannot participate due to medical exclusions (other needed medications, high liver enzymes, history of melanoma, etc.) To date, the drug has proven non-toxic and there have been essentially no dropouts in this study to date. The promise of a new treatment depends on you.
Call the APF on 866-APF-3635 or 301-347-7166.
We will connect you with a Research Coordinator that will answer all your Questions and concerns. We will work to make this process as easy as possible for you. It is 5 months of your life in order to give a future to ALL with EPP! To date there is no public information on the future of Scenesse. If we are able to get two FDA-approved treatments on the market, it may lower costs. If we do not do all we can as a community, we will have no treatment of EPP.
Bottom line: We need YOU! Please dig deep and consider being part of this changing moment for all with EPP.
Pet Calendar Update!
Thank you all for submitting your pets for the 2019 Pet Calendar Contest.
We have had an overwhelming response of submissions from our community. Please note that we are working diligently to have this calendar ready for you to purchase soon. Stay tuned for more details!
RSVP Today!
Patient Education and Support Meeting
LOCATION
Double Tree Hotel - San Diego - Hotel Circle
DATE AND TIME
12/01/18 6:00pm - 12/01/18 8:00pm
1515 Hotel Circle South San Diego, CA 92108 Room:Cortez Family and Friends are welcome to attend. We look forward to seeing you there.
Contact Information
Is your contact information up to date?
If not please give us a call @ 866-APF-3635 or Email to porphyrus@porphyriafoundation.com
American Porphyria Foundation 1.866.APF.3635 porphyriafoundation.org
STAY CONNECTED
"Remember.Research is the Key to Your Cure!"
Michelle Bridges AIP
Wednesday - November 7, 2018 @ 17:56:13
I spent my whole life with unexplained symptoms. Sometimes my legs would give out, and I would have back pain, abdominal pain, slurred speech, difficulties walking and so forth. When I was 16, doctors found a mass in my brain. After removing what they could, all my health problems were blamed on post surgical changes.
In 2005, the doctors began to wonder about other causes. I was sent to see a Gastroenteritis and diagnosed with pancreatitis. But there was no explanation for my abdominal pains that would come out of nowhere and then disappear. The doctors decided that is was due to a problem with my digestion and cut part of my bile duct. However, I landed back in the hospital with pancreatitis. Then nothing happened for about eight months.
At the end of July 2006, I began to have problems again. They put me on Reglan with the intention of running more tests and after about a week, I woke up feeling bizarre. I was in a haze and unable to function. I had called a friend and asked her to take me to the hospital. I turned to walk down the hall towards my bedroom so I could lie down, but my legs would not move. I remember standing there thinking you bend the knee and extend the leg. By the time I had dragged myself down the hall, my arms wouldn't cooperate and I fell over.
When the paramedics got the house I swear I asked if I was having a stroke. My son says I just lay there with my tongue hanging out, in and out of consciousness and seizure activity. I don't remember. The ER released me to my husband with the diagnosis allergic reaction. The following day I could not speak. Again, I went to the ER, but this time it was to a different hospital, one covered through our insurance. The doctor told my husband I was being over-medicated, and then a student at the hospital noticed the color of my urine and suggested to the doctor that my condition might be porphyria. She was correct!
I don't remember anything from the end of 2006 to 2008. Treatment for my symptoms made some things worse. I had been active in my church and home schooled other peoples children as well as my own, but I had to stop educating others and withdrew from some activities at church.
We have had a hard time finding doctors who will treat me. This disease is rare and doctors aren't experienced with it. With my new oncologists gentle touch and the support of the staff at our new hospital, I have become more active. I am not spending as much time in the hospital, and I still home school my children, write curriculum and resource guides, and do crafts for craft fairs and whatever else I can to support the church and help others. I do all of these activities from my bed because I am too weak and have too much pain to leave the bedroom for long stretches and I get sick so easily. But I do like to have some level of activity in my life and so I try to contribute where I can while being careful of my health.
Porphyria Post
Wednesday - October 31, 2018 @ 16:27:14
Porphyria Post
Last Day to Vote!
Tomorrow is the last day to capture votes for your furry friends! We look forward to sharing the results with our community.
If you haven't already done so, please go to the APF website to see everyone's submissions!
Make sure that you see your share this link and your individual voting pages with your family and friends.
The final 12-month wall calendar will feature your wonderful animals, porphyria facts, tips, and important dates. Voting for the top slots will be held online and will benefit our critical Protect the Future program.
·ALL photo entries will be included somewhere in the calendar, though the top dogs will be featured on the cover and in each month!
·Starting October 23rd - Your pets photo will be posted on the APF website for all to see. Youll be able to share the link with family and friends to encourage a vote! Anyone can participate with a $1+ donation per vote.
Important Dates:
·Voting: October 23-November 8
·Calendars ready for sale and shipping on November 25th!
Are you ready to be a medical hero? A new clinical trial with an oral investigational drug intended to reduce phototoxicity in people living with EPP has begun. Dont delay!
Pharmaceutical company: Mitsubishi Tanabe
Length on treatment: 16 weeks
Treatment: Oral (pills)
Number of appointments: 7-9
Arrangements: Includes Travel/hotel/food
Stipend: Each participant will receive a stipend
Locations: New York City, NY/Winston-Salem, NC/Miami, FL/Galveston, TX/Salt Lake City, UT/San Francisco, CA
How many participants do we need: 102!!!
Please contact the APF at 1.866.APF.3635 if you are interested in participating.
"RememberResearch is the key to YOUR cure!"
Each Step Toward Finding an Effective Treatment is Important!
Website Update!
You are now able to access the new website from any browser and on any device at www.porphyriafoundation.org
Thank you to our members, industry partners, and our brilliant physicians for making this update possible!
RSVP TODAY!
Patient Education and Support Meetings coming your way. Contact the APF today to RSVP! We look forward to seeing you there.
Patient Education and Support Meeting - San Francisco
LOCATION UCSF Medical Center 505 Parnassus Ave. Room: S-157 San Francisco, CA 94143
DATE AND TIME 11/11/18 5:30pm - 11/11/18 7:00pm
Seating is limited. Please RSVP Today!!
Patient Education and Support Meeting - San Diego
LOCATION Double Tree Hotel - San Diego - Hotel Circle 1515 Hotel Circle South San Diego, CA 92108
DATE AND TIME 11/11/18 5:30pm - 11/11/18 7:00pm
Room: SOLANA Please RSVP Today! Please note that there will be a Porphyria Specialist on site. We look forward to seeing you there!
We are excited to announce that we will launch our newly updated APF website TOMORROW. It is optimized to offer the patient community and healthcare professionals easy access to critical content - written by renowned experts - on all of the porphyrias.
On Thursday, November 1, 2018 you will be able to access the new website from any browser and on any device at
Thank you to our members, industry partners, and our brilliant physicians for making this update possible!
Mobile view of the New APF Homepage!
Pet Calendar Update
CALENDAR UPDATE: We are a week into voting and have seen an outstanding response! There is still a week left so remember to vote for your favorite pet picture and show your support of the APF. You can visit the APF website and see all of our amazing entries to pick who you want to see on the cover of the 2019 APF Pet Calendar! All photos will be included in the calendar though. Happy voting!
Important Dates:
Voting: October 23rd - November 8th
Calendars ready for sale and shipping on November 25th!
Starting your holiday shopping? Many of you purchase numerous items from Amazon. Did you know that your you can support the APF through the AmazonSmile program! Amazon donates 0.5% of the price of your eligible AmazonSmile purchases to the charitable organization of your choice.
Please make the APF your choice of a charitable donation. Support porphyria while shopping!
Note, this program to provide donations to the APF will ONLY be available to shoppers who visit Amazon via a special web address, namely, www.smile.amazon.com instead of the normal www.amazon.com homepage.
It is easy and free! AmazonSmile is the same Amazon you know - same products, same prices, same service.
VOTE: 2019 Pet Calendar, RSVP - Patient Education Meetings and EPP Clinical Trials
Let the voting begin!
Submissions have now closed for the 2019 Pet Calendar Contest. We appreciate all of your entries and can't wait to see how well everyone does in the contest.
If you haven't already done so, please go to the APF website to see everyone's submissions!
Make sure that you see your share this link and your individual voting pages with your family and friends.
The final 12-month wall calendar will feature your wonderful animals, porphyria facts, tips, and important dates. Voting for the top slots will be held online and will benefit our critical Protect the Future program.
ALL photo entries will be included somewhere in the calendar, though the top dogs will be featured on the cover and in each month!
Starting October 23rd - Your pets photo will be posted on the APF website for all to see. Youll be able to share the link with family and friends to encourage a vote! Anyone can participate with a $1+ donation per vote.
Important Dates:
Voting: October 23-November 8
Calendars ready for sale and shipping on November 25th!
Are you ready to be a medical hero? A new clinical trial with an oral investigational drug intended to reduce phototoxicity in people living with EPP has begun. Dont delay!
Pharmaceutical company: Mitsubishi Tanabe
Length on treatment: 16 weeks
Treatment: Oral (pills)
Number of appointments: 7-9
Arrangements: Includes Travel/hotel/food
Stipend: Each participant will receive a stipend
Locations: New York City, NY/Winston-Salem, NC/Miami, FL/Galveston, TX/Salt Lake City, UT/San Francisco, CA
How many participants do we need: 102!!!
Please contact the APF at 1.866.APF.3635 if you are interested in participating.
"RememberResearch is the key to YOUR cure!"
Each Step Toward Finding an Effective Treatment is Important!
New Publication - Journal of Pediatrics
Dr. Manisha Balwani, Porphyria Expert at Mount Sinai in New York City recently authored a publication that was recently released in the Journal of Pediatrics.
Patient Education and Support Meetings coming your way. Contact the APF today to RSVP! We look forward to seeing you there.
Patient Education and Support Meeting - Missouri
LOCATION THIS MEETING HAS BEEN CANCELLED
DATE AND TIME 10/27/18 12:00pm - 10/27/18 2:00pm
This meeting has been cancelled and will be rescheduled at a later date. If you have any questions, please reach out to Edrin at the APF!
Patient Education and Support Meeting - San Francisco
LOCATION UCSF Medical Center 505 Parnassus Ave. Room: S-157 San Francisco, CA 94143
DATE AND TIME 11/11/18 5:30pm - 11/11/18 7:00pm
Seating is limited. Please RSVP Today!!
Patient Education and Support Meeting - San Diego
LOCATION Double Tree Hotel - San Diego - Hotel Circle 1515 Hotel Circle South San Diego, CA 92108
DATE AND TIME 11/11/18 5:30pm - 11/11/18 7:00pm
Room: SOLANA Please RSVP Today! Please note that there will be a Porphyria Specialist on site. We look forward to seeing you there!
Emergency Room Guidelines for Acute Porphyrias
Thursday - October 25, 2018 @ 22:19:29
Emergency Room Guidelines for Acute Porphyrias
These Emergency Room Guidelines (download PDF) cover essential information for the emergency physician treating a patient in an acute porphyria attack, including common precipitating factors, typical presentation and other diagnostic clues, making the initial diagnosis, common sequelae and best practices for treatment. A PowerPoint presentation (as PDF) for instruction is also available.
Please note: These Guidelines are for Physician Use Only. The APF sells a separate, personalized Emergency Room and Primary Care Physician Kit that contains all the information acute porphyria patients need to have with them in the Emergency Room (medical journal articles, information about diagnostic labs and Panhematin, room for your own diagnostic test results).
Neville R Pimstone MD, PhD, Head Liver Diseases Greater West Los Angeles Veterans Affairs, Professor Emeritus UC Davis Karl E. Anderson MD, Professor, Departments of Preventive Medicine and Community Health, Internal Medicine, Pharmacology and Toxicology; Associate Director, General Clinical Research Center, Director, Porphyria Center and Laboratory, University of Texas Medical Branch, Galveston, Texas Bradley Freilich, MD, Kansas City Gastroenterology and Hepatology, LLC
KEY POINTS
1. The human porphyrias are clinical disorders reflecting defects in heme biosynthesis. 2. Acute porphyrias cause acute attacks of neurological symptoms that can be life-threatening. 3. Acute attacks are triggered by certain drugs, sex steroid hormones, reduced intake of calories and carbohydrate, alcohol and unknown factors. 4. Many of these factors stimulate heme synthesis in the liver, which in the face of a metabolic enzyme defect, leads to increased production of heme precursors that may be neurotoxic. 5. Delta-aminolevulinic acid (ALA) and porphobilinogen (PBG), are porphyrin precursors and intermediates in the heme biosynthetic pathway. 6. ALA and porphobilinogen (PBG) are almost always elevated in urine during an acute attack of porphyria. 7. The most common emergency room (ER) clinical presentation is acute abdominal pain. Other features may include seizures, confusion and hallucinations, and a progressive polyaxonal motor neuropathy, which can progress to paralysis and respiratory failure requiring a ventilator. 8. A high index of suspicion in the presence of nonspecific symptoms is important for diagnosis. A family history of porphyria, female sex, onset during the luteal phase of the menstrual cycle, or recent use of a porphyrinogenic drug may be diagnostic clues. 9. A new diagnosis of porphyria as the cause of acute symptoms must be substantiated by finding a substantial increase in urine porphobilinogen (PBG). 10. Treatment should start promptly after the diagnosis is made. Mild attacks are sometimes treated with glucose loading (e.g. 3L of 10% glucose daily by vein). 11. Most acute attacks should be treated with hemin (Panhematin, Recordati Rare Diseases at: www.recordatirarediseases.com or 866-654-0539) 3-4mg/kg into a large peripheral vein or venous access port daily for 4 days. Reconstituting Panhematin with human serum albumin rather than sterile water is recommended prior to infusion. This helps prevent phlebitis at the site of intravenous infusion. 12. Hospitalization is usually required for symptomatic treatment of pain, nausea and vomiting, correction of electrolyte imbalance and observation for respiratory impairment, either to a general medical service or ICU.
Pediatrics & EPP
Wednesday - October 24, 2018 @ 16:00:08
Share your article!
Dear Dr. Balwani,
We are pleased to let you know that the final version of your article Diagnostic Delay in Erythropoietic Protoporphyria is now available online, containing full bibliographic details.
To help you access and share this work, we have created a Share Link a personalized URL providing 50 days' free access to your article. Anyone clicking on this link before December 11, 2018 will be taken directly to the final version of your article on ScienceDirect, which they are welcome to read or download. No sign up, registration or fees are required.
Understanding the thoughts and feelings of other individuals is essential for navigating the social world. But empathy is a complex process, based in part on fleeting facial expressions. Research suggests that we empathize by effectively putting ourselves in others shoes: for example, when we observe someone feeling sad, we simulate their experience by activating the same regions of the brain that are involved when we feel sad ourselves.
A study in the Journal of Neuroscience in February bolsters this idea using rare individuals with mirror-touch synesthesia. When watching another individual being touched, these people actually feel a touch on the same part of their own body. Neuroscientist Michael Banissy and his colleagues at University College London tested whether this heightened ability to simulate another persons experience would cause eight mirror-touch synesthetes to excel at recognizing the emotions embedded in facial expressions. They did, correctly identifying 92 percent of the facial expressions tested compared with the 81 percent identified by control subjects. Their success probably stemmed from their simulation expertise rather than a general agility with faces because further experiments showed they were no better than controls at recognizing a persons identity.
For the rest of us without mirror-touch synÂesthesia, the simulation process is the same but less pronounced, Banissy says. So the next time you find yourself sympathizing with someone who looks sad, thank the part of your brain that feels you frown.
I enjoyed reading this over, when we face trials, illness, over outward expressions can speak volumes and usually we say were doing ok or good! So be honest with how you feel!
Thanks to Michele for letting me pass this on to you.
Do you have PCT? Are you interested in participating in research? Do you have Hepatitis C? If you answered YES to these questions, this is for you.
We need YOU for a clinical trial!
The purpose of this clinical trial is to assess whether Harvoni alone is an effective therapy in active PCT patients with Chronic Hepatitis C.
Who can participate?
â?¢ Adult patients with PCT who also have Hepatitis C
If you are interested in participating please contact Edrin Williams, Director of Patient Services at the APF office at 301.347.7166 for additional information.
WE NEED YOU FOR THE EPP CLINICAL TRIAL! IF NOT YOU, THEN WHO??
1. Do you have a diagnosis of EPP? 2. Are you interested in participating in research?
If so, we NEED you!
Participants are desperately needed for participation in a Phase II clinical trial to study an investigational drug for EPP. CONTACT THE APF ON 1-866-APF-3635 or email porphyrus@porphyriafoundation.com to learn more.
An oral investigational drug has been developed with the potential to increase sunlight duration and tolerance in individuals with EPP. This phase II clinical trial is needed to understand if the investigational drug works and to ensure that it is safe. The study duration is 24 weeks including follow-up. You must be age 18-70 to participate and have a confirmed diagnosis of EPP. All travel is included and will be arranged by a concierge service.
â?¢ We need patients at all participating sites (NYC, Wake Forest, Miami, Utah and UCSF) â?¢ Please note that the site at the University of Utah have just opened for enrollment. â?¢ The site at the University of California San Francisco will be enrolling patients soon.
Contact the APF today! 1-866-APF-3635.
"RememberResearch is the key to YOUR cure!" Each Step Toward Finding an Effective Treatment is Important!
Pet Calendar Update! Thank you all for submitting your pets for the 2019 Pet Calendar Contest. Submissions are due by Sunday, October 21, 2018. See the instructions below on how you can get involved.
We have had an overwhelming response of submissions from our community. Please note that we are working diligently to upload your pet photos to the APF Website. Please be on the lookout for updates from the APF team regarding your profile.
CALLING ALL PETS!!! Welcome to the 2019 APF Pet Calendar Contest YOUR pet can win a coveted spot on this fun and informational calendar! The final 12-month wall calendar will feature your wonderful animals, porphyria facts, tips, and important dates. Voting for the top slots will be held online and will benefit our critical Protect the Future program. Enter your pets then a link will be created to encourage voting! Pets are an important part of our lives and provide much-needed unconditional love in tough times. We look forward to showing off your special friends as your photos roll in!!
Heres how it works:
To Enter (free!): â?¢ Email a picture of your pet(s) to the APF by October 21st. â?¢ Send to autumnlee@porphyriafoundation.org with the subject heading APF Pet Calendar. Make sure to include: â?¢ Pet type and name â?¢ Owner name, email, and mailing address** â?¢ Send a caption, too! (optional)
To Vote: â?¢ ALL photo entries will be included somewhere in the calendar, though the top dogs will be featured on the cover and in each month! â?¢ Starting October 17th - Your pets photo will be posted on the APF website for all to see. Youll be able to share the link with family and friends to encourage a vote! Anyone can participate with a $1+ donation per vote.
â?¢ You may enter one photo that contains as many of your pets you wish to include. Remember that this photo will be voted on so choose your best! â?¢ Anyone can enter â?¢ No professional or copyrighted photos â?¢ Any type of pet can be submitted; past and present â?¢ No humans please â?¢ Photos cannot be photo shopped or professionally edited
Important Dates:
â?¢ Submissions due by October 21st â?¢ Voting: October 23-November 8 â?¢ Calendars ready for sale and shipping on November 25th!
Thank you to Nicole Castellano for leading this effort for the APF!
Please contact Autumnlee at the APF office with any questions at: autumnlee@porphyriafoundation.org or call (301) 347-7166.
GOOD LUCK TO EVERYONE and HAVE FUN!
Get Involved
â?¢ Research â?¢
â?¦ Donate â?¦
â?¢ Visit our Website â?¢
Contact Information
Is your contact information up to date? If not please give us a call @ 866-APF-3635 or Email to porphyrus@porphyriafoundation.com
Welcome our newest Protect The Future Doctor Amy Dickey. She will be training with our Expert Dr. Karl Anderson @ UTMB and she will have other mentors. Please give her a warm welcome and watch her presentation below:
Last week I was able to teach on porphyria to the internal medicine residents at Massachusetts General Hospital. Below is a link with the video if youre interested in hearing it. Its a talk to physicians, so be prepared for lots of medical jargon â?º
Im a physician at MGH trained in internal medicine, pulmonary, and critical care, and I have EPP. Padmini Pillai who shares at the end is a post-doc in biomedical engineering at MIT, and she has AIP.
#porphyria#awareness
Treatment and Management of EPP
Saturday - October 13, 2018 @ 23:34:51
Treatment and Management
1. Sunlight protection
Protection from sunlight is the mainstay of management of EPP, and this is necessary throughout life. Disease severity and porphyrin levels in erythrocytes and plasma probably remain high and relatively constant throughout life in EPP. However, this has been little studied and more longitudinal observations are needed. Life style, employment, travel and recreation require adjustment in order to avoid painful reactions to sunlight and even from exposure to fluorescent lighting. For these reasons EPP can substantially affect quality of life.
Protective clothing, including broad-brimmed hats, long sleeves, gloves and trousers (rather than shorts), is beneficial. Several manufacturers specialize on clothing made of closely woven fabrics for people with photosensitivity.
2. Other considerations
In an occasional patient, protoporphyrin causes liver problems, so monitoring liver function is important. EPP patients should also not use any drug or anesthetic which causes cholestasis (slowing down bile flow), and should also avoid alcohol. Women should avoid medications containing estrogen (birth-control pills, hormone replacement therapy), and men should avoid testosterone supplements, as these substances can also have deleterious effects on the liver of a person with EPP.
Consult a specialist. Because EPP is a rare condition, most physicians are not knowledgeable about it. Contact The American Porphyria Foundation, 713-266-9617 for contact with an expert and to provide further information. A Medic Alert bracelet with instructions to contact a specialist if needed is a worthwhile precaution.
Yearly monitoring. Testing to include erythrocyte total protoporphyrin, plasma porphyrin, complete blood counts, ferritin and liver function tests should be done yearly. Porphyry levels are expected to be stable and liver tests to remain normal. EPP patients may have evidence of iron deficiency, and an iron supplement may be advisable if the serum ferritin is below about 20 ng/mL.
Vitamin D. Because they avoid sunlight, EPP patients are likely to be deficient in vitamin D. A vitamin D supplement with calcium is recommended for bone health.
Liver protection. It is important to avoid other causes of liver disease that might promote the development of liver complications from EPP. Patients should avoid alcohol and other substances that might damage the liver, including many herbal preparations, and be vaccinated for hepatitis A and B.
Surgical lights. Strong operating room lights can cause photosensitivity of the skin and even surfaces of internal organs. Flexible membrane filters, such as CL5-200-X from Madico Co., are available to cover surgical lights and offer some protection. This is especially important in EPP patients with liver failure, which causes even greater increases in protoporphyrin levels and photosensitivity.
Drugs. Drugs that are harmful in other porphyrias are not known to make EPP worse, but are best avoided as a precaution. This may include estrogens and other drugs that might reduce bile formation. A short course of a non-steroidal anti-inflammatory drug can provide some pain relief after an episode of photosensitivity, but can cause ulcerations of the digestive track especially with prolonged use.
Laser treatment. According to Dr. Roth, laser treatments for hair removal or eye surgery have not been a problem in EPP people. But the doctor should be made aware of the diagnosis, and that laser output between 400 and 650 nanometers might be harmful. Before hair removal treatment, the doctor may irradiate a small area of the skin to be treated for the length of time it will take to do the hair removal to ascertain if the patient would react within the period of time that a reaction to sunlight would be expected in that patient.
CALLING ALL PETS!!!
Thursday - October 11, 2018 @ 06:30:03
CALLING ALL PETS!!!
Welcome to the 2019 APF Pet Calendar Contest YOUR pet can win a coveted spot on this fun and informational calendar! The final 12-month wall calendar will feature your wonderful animals, porphyria facts, tips, and important dates. Voting for the top slots will be held online and will benefit our critical Protect the Future program. Enter your pets then a link will be created to encourage voting! Pets are an important part of our lives and provide much-needed unconditional love in tough times. We look forward to showing off your special friends as your photos roll in!!
Heres how it works:
To Enter (free!): â?¢ Email a picture of your pet(s) to the APF by October 21st. â?¢ Send to autumnlee@porphyriafoundation.org with the subject heading APF Pet Calendar. Make sure to include: â?¢ Pet type and name â?¢ Owner name, email, and mailing address** â?¢ Send a caption, too! (optional)
To Vote: â?¢ ALL photo entries will be included somewhere in the calendar, though the top dogs will be featured on the cover and in each month! â?¢ Your pets photo will be posted on the APF website for all to see. Youll be able to share the link with family and friends to encourage a vote! Anyone can participate with a $1+ donation per vote.
â?¢ You may enter one photo that contains as many of your pets you wish to include. Remember that this photo will be voted on so choose your best! â?¢ Anyone can enter â?¢ No professional or copyrighted photos â?¢ Any type of pet can be submitted; past and present â?¢ No humans please â?¢ Photos cannot be photo shopped or professionally edited
Important Dates:
â?¢ Submissions due by October 21st â?¢ Voting: October 23rd-November 8th â?¢ Calendars ready for sale and shipping on November 25th!
Thank you to Nicole Castellano for leading this effort for the APF!
Please contact Autumnlee at the APF office with any questions at: autumnlee@porphyriafoundation.org or call (301) 347-7166.
GOOD LUCK TO EVERYONE and HAVE FUN! Click below to watch Nicole's Video Introduction!
Recordati Rare Diseases is launching a new 350mg dosage strength of Panhematin
Recordati Rare Diseases is launching a new 350mg dosage strength of Panhematin. This is the first significant change to Panhematin since it was approved over 30 years ago. The prescribing instructions have numerous updates including the removal of the boxed warning, addition of clinical studies section, updates to the adverse events section, and changes to dosing and administration. The distribution process for Panhematin will stay the same.
The new dosage strength is being launched to better accommodate the current weight of patients and the current dosing of Panhematin in clinical practice.
A new copay assistance program for Panhematin will be available starting in July 2017. This program can help eligible patients with their copay when they receive Panhematin on an outpatient basis. Please see the Panhematin website (www.panhematin.com) for more information.
What is Variegate Porphyria?
Tuesday - October 2, 2018 @ 06:30:14
Variegate Porphyria (VP)
What is Variegate Porphyria?
VP is caused by a mutation in the enzyme protoporphyrinogen oxidase (PPOX), which is part of the pathway that produces porphyrins and heme. Acute attacks are similar to those in AIP and HCP but are unusual. A more common sign of the disease is blistering skin lesions, which are chronic in many people with VP.
Acute attacks almost always start with severe pain in the abdomen but sometimes in the chest, back, or thighs, and are often accompanied by nausea, vomiting, and constipation. Heart rate and blood pressure are commonly increased. These symptoms and signs are all due to the effects of the disease on the nervous system. Confusion, convulsions, and muscular weakness, due to impairment of the nerves controlling the muscles, may lead to paralysis. An acute attack usually lasts for days or weeks. Recovery from severe paralysis is generally slow.
Who gets Variegate Porphyria?
VP is especially common in South Africa in individuals of Dutch ancestry, where it has been estimated that 3 in 1,000 of the white population are affected. It is much less prevalent in other countries. Like AIP and HCP, it is an autosomal dominant disorder, meaning that a mutation is present in only one of the pair of PPOX genes.
What causes Variegate Porphyria?
As in HCP, acute attacks of VP are unusual except in the presence of environmental activating factors, such as drugs, hormones, and dietary changes. See HCP for discussion of these issues.
How is Variegate Porphyria Diagnosed?
Urine ALA and PBG are increased during attacks, but as in HCP, these may increase less and decrease more rapidly than in AIP. Plasma porphyrins are frequently increased in VP, in contrast to AIP and HCP, and the plasma of VP patients displays a distinctive fluorescence peak, which is diagnostic. Fecal porphyrins are also elevated and are predominantly coproporphyrin III and protoporphyrin.
Management and prevention are the same as in AIP and HCP. Blistering skin lesions are much more common than in HCP and are not readily treated. The only effective preventive measure is use of protective clothing.
What is the long-term outlook after an attack of Vairegate Porphyria?
The prognosis is usually good if the disease is recognized and treated promptly, before nerve damage develops. Although symptoms usually resolve after an attack, recovery of neuromuscular function (in a severe case) may require several months. Mental symptoms may occur during attacks but are not chronic. Premenstrual attacks often resolve quickly with the onset of menses.
Can attacks be prevented?
Yes, particularly with regard to drugs and diet. Genetic VP carriers should become informed on medications to avoid (see information on AIP and HCP) and should be prepared to point their healthcare providers to on-line drug lists that are regularly updated. The American Porphyria Foundation offers a mobile phone app that pulls up this information on line (http://porphyriadrugs.com/). A Medic Alert bracelet is useful for a situation in which the patient is incapacitated. Very frequent premenstrual attacks can be prevented by a gonadotropin-releasing hormone (GnRH) analogue (Lupron, Zoladex, others) administered with expert guidance. In selected cases, frequent noncyclic attacks can be prevented by once- or twice-weekly infusions of hemin.
Individuals who are prone to attacks should consume a normal balanced diet. Despite on-line discussion, there is no evidence that pushing carbohydrate prevents attacks, and it has the side effect of weight gain, which is undesirable for most people. Fasting, fad diets (for example, high protein) and gastric reduction surgery should be avoided. If weight loss is desired, it is advisable to consult a physician and a dietitian about an individualized diet with modest caloric restriction (ca. 10%), which will produce gradual weight loss without increasing the risk of an attack of porphyria. Exercise is safe in porphyria, and recommended.
Porphyria Post
Monday - October 1, 2018 @ 06:30:10
Porphyria Post
CLINUVEL INITIATES VARIEGATE PORPHYRIA PROOF OF CONCEPT STUDY
·Today CLINUVEL announced it has initiated a new clinical trial in variegate porphyria (VP), set to commence in Europe in 2019.
***ANY PURCHASE THIS WEEK WILL RECEIVE FREE APF Gratis***
APF Merchandise for Sale!
Our hope is that you all will buy and wear APF merchandise to help promote and raise awareness about Porphyria. Please note that all orders will go through APF employee Amy Chapman. Amy@porphyriafoundation.org
****We DO NOT accept orders through FB****
Please submit your order and payment toAmy@porphyriafoundation.org or call 1-616-213-0030 directly and leave a detailed message.
Please send an email and include the following: Full Name, complete mailing address, phone number.
Order: Please send your items that you would like to order by quantity & size for each T-shirt. For privacy purposes, please send in the email either the Visa/MC Number, Expiry Date, & CVV code on the back of your card. If you prefer I can contact you by phone.NO CC info is EVER kept on file!
All orders include shipping prices for USPS only **International orders may be priced higher allow 48 hours**
Please review this link for color and size selection: http://www.porphyriafoundation.com/content/apf-merchandise
ITEMS FOR SALE:
BOGO - Buy One Get One Free!
V-Neck Short Sleeve Shirts - Deep Purple
Sizes available: Adults Only- S, M , LG, X-LG, 2XL, 3XL- (X-LG. 2XL, 3XL are $3.00 each more)
Price Each- S, M, L $20.00 Each ** X-LG. 2XL, 3XL are $3.00 each more**
Long Sleeve Shirts~ are 6 ounces each 100% Cotton. Colors are Medium Gray or Purple
Sizes available Adults Only- S, M , LG, X-LG, 2XL, 3XL- (X-LG. 2XL, 3XL are $3.00 each more)
Price Each- S, M, L $20.00 Each ** X-LG. 2XL, 3XL are $3.00 each more**
Long Sleeve 100% Cotton Thick Zip Up Hoodie Colors are Medium Gray or Royal Purple.
Price Each- S, M, L $45.00 Each ** X-LG. 2XL, 3XL are $5.00 each more**
Long Sleeve 100% Cotton Pull Over Hoodie Colors are Medium Gray or Purple
Price Each- S, M, L $40.00 Each ** X-LG. 2XL, 3XL are $5.00 each more**
Gray Soft Baseball cloth hats adjustable hat backside- $22.00 Each (Fits Adults Sizes)
Wristbands- 2/$6.00 or $3.00 each Colors are Blue or Purple
Porphyria Car or Fridge Magnet $8.00 each
September is Pain Awareness Month
Friday - September 28, 2018 @ 06:00:03
September is Pain Awareness Month
The month of September has been declared Pain Awareness Month. Pain Awareness Month is a time when various organizations work to raise public awareness of issues in the area of pain and pain management.
The first Pain Awareness Month was in 2001, when the ACPA led a coalition of groups to establish September as Pain Awareness Month. ACPA established Partners for Understanding Pain and 80 organizations, both health care professionals and consumer groups, including the NAACP supported the effort.
The key to raising awareness is to get involved. There are many things that you can do to help promote Pain Awareness Month.
·Talk with Friends & Family: Let them know that September is Pain Awareness Month. Like the ACPA on Facebook. Encourage your friends to do the same.
·Talk with your Healthcare Provider: Let them know that September is Pain Awareness Month. You also can share the tools to better communicate with your healthcare team found at the links below:
·Call your local government and community leaders to let them know about Pain Awareness Month and issues of pain and pain management
·Call your local media and ask them if they are doing a story on Pain Awareness Month.
·Donate to the APF: Your contributions allow us to help fulfill our mission and work year-round to raise awareness and support for those with chronic pain. www.Porphyriafoundation.org
·Take care of yourself! Take time out for yourself this month.
NEW! Member story of Jeannine Keith HCP
Thursday - September 27, 2018 @ 06:30:06
What is your name?Jeannine Keith
Type of Porphyria?Hereditary Coproporphyria (HCP)
When & how long before you were diagnosed? I started having symptoms in March of 2010 and got my formal diagnosis June of 2010. In May of 2010 my Gastroenterology Dr. Peter Heit sent me for a urinalysis of the porphyrin levels in my body after I had every other organ tested with no answers to my pain. When the urinalysis can back positive he found me my savior Dr. Bari.
Who was able to diagnose you?Name/place? Dr. Fazal Bari, Oncology and Hematology Specialists
What has been your experience with porphyria? It is an unknowing disorder and can sneak up on you at any time day or night. I have had more good days than bad thankfully.
What symptoms do you have and how long do they last for? I have dealt with pain in my side, back, and chest. I have also had experienced nausea, neuropathy, and suffer from anxiety.
What treatment do you get?How often do you have to get treatment? Since 2010 I have been doing IV therapy of D5W monthly. When I started this, I was going to my doctors office 3 days out of the month during my menstrual cycle and spent 4 hours getting my hydration in the office. After doing this for a few months and knowing that this type of treatment was working for me I got a PICC line. I had that for 6 weeks and was able to have a nurse come to my house and I could run my IVs from home. After I had the PICC line taken out, I had my first port in 2011. I had my port in for 2 years before opting to have it moved from its original position. I have had my second port since November 2012 and still do my IV therapy from home, work or wherever I am.
Do you think a new medication would help you? What I am doing has been helping me for the past 8 years. I am not sure if new medication would help me.
How does having porphyria affect your daily activities? It really doesnt affect my daily activities unless I am having an attack. I live my life the way any person would, and I make sure I stay hydrated and attack free as best I can.
How has porphyria affected your life & family living? It is my way of living and I try not to let it affect my life or family living. I have this disorder, but I am not this disorder.
What are the positives of knowing you have a rare disorder? The positives are that when I have pain, nausea, vomiting or an anxiety attack I know what could most likely be the cause. I can also educate others about porphyria.
How do you keep yourself encouraged from day to day? I think about all the positives in my life and know that I am blessed. My 4-year-old son Ayden and my husband Sean are also my loves, my support and my heart.
If you could give an advice to another one suffering what would you say? Dont let this disorder define who you are. You are strong and can fight this. I know having this disorder is terrible and we all have our good days and our bad days, but we have to keep our heads held high and smile because some days that is all that might get us through. Remember, you never know what someone is going through, and I like many, smile and say everything is ok but sometimes it is ok to be truthful and say when it is not ok.
How has the APF been there to assist you?(Materials, videos, calls, FB) There website has many articles, blogs and useful information. I have met so many people through groups on FB and they are wonderful!
What else would you like to mention? I am very lucky to have an amazing support system of Family, friends and medical professionals. I want to thank my husband Sean for always being by my side, my son Ayden for always making me laugh & My parents for always being there for me through learning about this disorder and getting answers. I also want to give a huge thank you to Dr. Bari for giving me my life back because without you I dont know where I would be with this disorder.
Thank you Jeannine Keith for sharing your inspiring journey with HCP! You always encourage us.
Baby Harmoni's Diagnosis of CEP Congenital Erythropoietic Porphyria.
Wednesday - September 26, 2018 @ 18:17:43
Last year in October 2017, Me and my fiance found out we were pregnant with our first child & we could not have been more excited. After waiting so long at 38 weeks our sweet little girl Harmoni Grace was here weighing 8 pounds 1 oz and twenty one and a half inches long.
A short few hours after Harmoni was born she had what the nurses thought was what they call a brick dust diaper, which is normal in babies just not nearly as dark as Harmonis was or the quantity.
We soon learned she was also born jaundice, so she was sent to the NICU to spend two days to help with the jaundice and to get to the bottom of the brick dust diaper. After two very long days and some testing doctors could not find anything wrong with Harmonis urine so we were cleared to head home.
As excited as we were to bring our baby girl home we just knew something wasn't right. For two whole months after that me and my fiance spent almost every day in Valley Children's Hospital trying to get to the bottom of all of her problems and symptoms.
Almost every time we took her in for vomiting, the red diaper, her being lethargic, and not eating they sent us home and told us we have a colic baby and we should hold her more and we would have to let her cry it out sometimes, that answer was never good enough for me. As a mother I knew something just wasn't right.Finally, after many trips to the emergency department we get admitted due to her hemoglobin being extremely low.
I can remember that day was a nightmare being questioned left and right and having a whole team of doctors come in to tell you your two-month-old has a hemoglobin that should be at her max limit which is a 13.6 but she is at the lowest a person should get a 7.0.
Doctors began to tell us that she will need a blood transfusion and we will be in the hospital until we can find the source of the bleed. This admission we spent almost two weeks in the hospital with the best doctors on our team to find some possible answers to what is going on with our baby girl.
On the fifth day the team of doctors came in, sat us down, & let us know they might have an answer to all of this madness going on with our sweet girl. They informed us they think Harmoni has Porphyria, and in that moment, I think we reacted in any way any parents would with hearing a big medical term without knowing any knowledge of what it is or what comes with it.
After a little more information from several different doctors we were able to take a deep breath and brace ourselves for whatever was to come next. The next few days consisted of meeting with a hematologist and genetics doctor and TONS of testing.
The results would take some time to come back.
We were finally discharged when harmonies hemoglobin was at a safe level. We were advised her levels would have to be checked every two weeks to make sure her hemoglobin stays at a safe level and to make sure she wont need another transfusion.
On September 6th she went in to test her levels and sure enough they were low again, so we were once again admitted for a transfusion and a Broviac Line placed. After two days Harmoni was safe enough to come home once again with the same routine of checking her blood levels every two weeks. On September 11th we got her results and it was confirmed she has Congenital Erythropoietic Porphyria.
Now, we are waiting for calls back to get into UCSF to meet with specialists to take the next step to helping her.
Even though she was diagnosed with porphyria will do everything in our power as her parents to make sure she in no way shape or form feels any different than others. And at only three months old our little girl is the strongest little girl I have ever met, we refuse to let this kick her butt, so we will always make sure she is Porphyria strong.
Thank you for sharing through a parents eye how special and wonderful your baby Harmoni is to you.Harmoni you are very special to us and how wonderful it is to have such loving and caring parents fighting for you.
To learn more about Congenital Erythropoietic Porphyria (CEP) please call our office at 1-866-APF-3635 or view our website www.porphyriafoundation.org
Porphyria Post
Tuesday - September 25, 2018 @ 06:30:00
Porphyria Post
Volunteers Needed!
The APF will be exhibiting at the two conventions this fall. Are you interested in volunteering at our booth? If so, please contact Edrin Williams, Director of Patient Services for additional information (edrinw@porphyriafoundation.org or 301.347.7166). This is an incredible opportunity to raise awareness about porphyria to those with little knowledge about this rare disease.
AASLD: San Francisco, CA
â?¢ Sunday, November 11 9:30 am - 3:00 pm time slots available
â?¢ Monday, November 12 9:30 am - 3:00 pm time slots available
ASH: San Diego, CA
â?¢ Saturday, December 11:00 a.m. - 5:00 p.m. time slots available
â?¢ Sunday, December 210:00 a.m. - 5:00 p.m. time slots available
â?¢ Monday, December 310:00 a.m. - 2:00 p.m. time slots available
EPP Clinical Trials
Are you ready to be a medical hero? A new clinical trial with an oral investigational drug intended to reduce phototoxicity in people living with EPP has begun. Dont delay contact the APF to learn more at 1-866-APF-3635 or porphyrus@porphyriafoundation.org
"RememberResearch is the key to YOUR cure!"
Each Step Toward Finding an Effective Treatment is Important!
Access to Care Toolkit for the Acute Porphyrias
A downloadable Access to Care Toolkit is a resource designed to help patients living with Acute Intermittent Porphyria (AIP), Hereditary Coproporphyria (HCP), and Variegate Porphyria (VP) or their caregivers, loved ones and healthcare providers secure access to Panhematin at their preferred health facility. We have recently learned of patients who are being denied this treatment from some hospitals and directed to secure another healthcare provider. If this has happened to you or someone you know or care for, please use these tools to request help from your state and local representatives and health advocacy organizations. We understand the debilitating effects of acute porphyria and hope these resources will help you secure access to Panhematin when you need it most.
The Toolkit contains the following materials:
â?¢ Healthcare Conversation Tracker is a simple form to record your conversations with doctors, insurance agents, etc.
â?¢ Customizable letter templates to record your details to use for doctors, state departments, insurance, etc.
â?¢ Access to Care Fact Sheet defines AIP, its symptoms and why it's important for patients to get immediate care
â?¢ Patient Bill of Rights can be used to support your appeal for access to treatment
This Toolkit can be found HERE. Contact the APF office today if you have questions!
Panhematin Study - Participants Needed!
Do you have AIP, VP or HCP? Are you interested in participating in research? If so, we are recruiting volunteers for the Panhematin Study.
Our researchers need people to volunteer for the Panhematin study. Completing this study will help prevent insurance companies from not paying for this treatment.
The purpose of this study is to determine if Panhematin is safe and effective for prevention of acute attacks of porphyria.
Please contact Edrin at the APF office (301.347.7166) if you are interested in participating.
Safe/Unsafe Drug Questionnaire for the Acute Porphyrias (AIP,VP,HCP & ADP) - VERY IMPORTANT
The APF is collaborating with researchers to identify new safe and unsafe drugs. We need your help. Are you experiencing adverse effects with any of your new medications? If so, please let us know in the questionnaire below how it affected you. We will share these results with our team of renowned Porphyria experts/researchers. They are in the process of updating our safe and unsafe drug list for the acute porphyrias. Your donations will help us educate physicians about the dangerous effects of unsafe drugs. Please click on the link below for the Questionnaire.
Safe and Unsafe Drug Questionnaire
RSVP TODAY!
Patient Education and Support Meetings coming your way. Contact the APF today to RSVP! We look forward to seeing you there.
Patient Education and Support Meeting - Cincinnati
We appreciate everyone who sent in their RSVP! We hope to see you on Saturday!!
See you on Saturday!
Patient Education and Support Meeting - Missouri
LOCATION
Mound Ridge Retreat and Mission Center 31 Agape Lane Cook Station, MO 65449
DATE AND TIME
10/27/18 12:00pm - 10/27/18 2:00pm
Question and Answer session by Porphyria Expert via Skype. Family and Friends are welcome to attend. Lunch will be provided.
I'll be there!
Maybe
I can't make it
Patient Education and Support Meeting - San Francisco
LOCATION
UCSF Medical Center 505 Parnassus Ave. Room: S-157 San Francisco, CA 94143
DATE AND TIME
11/11/18 5:30pm - 11/11/18 7:00pm
Seating is limited. Please RSVP Today!!
I'll be there!
Maybe
I can't make it
Get Involved
â?¢ Research â?¢
â?¦ Donate â?¦
â?¢ Visit our Website â?¢
Contact Information
Is your contact information up to date?
If not please give us a call @ 866-APF-3635 or Email to porphyrus@porphyriafoundation.com
American Porphyria Foundation 1.866.APF.3635 porphyriafoundation.org
STAY CONNECTED
"Remember.Research is the Key to Your Cure!"
Acute Intermittent Porphyria Story of Jessica Melton
Wednesday - September 19, 2018 @ 17:18:23
Jessica Melton
Acute Intermittent Porphyria
My name is Jessica and I have Acute Intermittent Porphyria {AIP}. I was unofficially diagnosed at nineteen years old, and then officially diagnosed via DNA testing at the age of twenty-five. By the time I was nineteen, I had already endured over ten surgeries, including several exploratory surgeries to try and find the root cause of my unexplained abdominal pain.
I was lucky, my hematologist started me on Panhematin. When the treatment was done, I could not believe it, my pain was gone for the first time in my life! I am now thirty-five and I still receive hematin on a regular basis.
Unfortunately, due to the chronic nature of the disease, more damage has been done to my nerves and organs. I was diagnosed with gastroparesis, neuropathic pseudo-obstruction, seizures, and Tourette syndrome. The icing on the cake is an extremely elevated ferritin level. This, we believe, was caused by the hematin. For two years now, I have been getting weekly inpatient 24-hour intravenous treatments ** to strip the iron from my body.
Despite the constant uphill battle with this disease, I was able to graduate with my associates degree in special education and my paralegal certificate. My life is certainly not perfect, but one thing is for sure; if I can overcome all of this, I can do pretty much anything I set my mind to.
Jessica Melton
Thank you, Jessica, for your courageous fight and determination to not allow Porphyria to overtake your joy for accomplishing your goals. Congratulations to you!
**If you would like to learn more about Acute Intermittent Porphyria or the Medications or Procedures used please contact the APF~ 1-866-APF-3635
Please talk with your physician before Starting/using/stopping any drug.
PORPHYRIA POST
Monday - September 17, 2018 @ 06:30:11
Porphyria Post
Welcome to the APF, Autumnlee! We are excited to welcome aboard our newest APF team member, Autumlee Meyer.
We are so excited to have you here with us and wish you nothing but the best in your future with the APF.
Please extend a warm welcome to Autumnlee! Email her at autumnlee@porphyriafoundation.org or give her a call at 301.347.7166 to say hello.
EPP Clinical Trials
Are you ready to be a medical hero? A new clinical trial with an oral investigational drug intended to reduce phototoxicity in people living with EPP has begun. Dont delay contact the APF to learn more at 1-866-APF-3635 or porphyrus@porphyriafoundation.org
"RememberResearch is the key to YOUR cure!" Each Step Toward Finding an Effective Treatment is Important!
Harvoni Study - PCT
Do you have PCT? Are you interested in participating in research? Do you have Hepatitis C? If you answered YES to these questions, this is for you.
We need YOU for a clinical trial!
The purpose of this clinical trial is to assess whether Harvoni alone is an effective therapy in active PCT patients with Chronic Hepatitis C.
Who can participate?
â?¢ Adult patients with PCT who also have Hepatitis C
If you are interested in participating please contact Edrin Williams, Director of Patient Services at the APF office at 301.347.7166 for additional information.
Panhematin Study - Participants Needed!
Do you have AIP, VP or HCP? Are you interested in participating in research? If so, we are recruiting volunteers for the Panhematin Study.
Our researchers need people to volunteer for the Panhematin study. Completing this study will help prevent insurance companies from not paying for this treatment.
The purpose of this study is to determine if Panhematin is safe and effective for prevention of acute attacks of porphyria.
Please contact Edrin at the APF office (301.347.7166) if you are interested in participating.
Safe/Unsafe Drug Questionnaire for the Acute Porphyrias (AIP,VP,HCP & ADP) - VERY IMPORTANT
The APF is collaborating with researchers to identify new safe and unsafe drugs. We need your help. Are you experiencing adverse effects with any of your new medications? If so, please let us know in the questionnaire below how it affected you. We will share these results with our team of renowned Porphyria experts/researchers. They are in the process of updating our safe and unsafe drug list for the acute porphyrias. Your donations will help us educate physicians about the dangerous effects of unsafe drugs. Please click on the link below for the Questionnaire.
Safe and Unsafe Drug Questionnaire
RSVP TODAY! Patient Education and Support Meetings coming your way. Contact the APF today to RSVP! We look forward to seeing you there.
Patient Education and Support Meeting - Cincinnati
LOCATION Cincinnati Children's Hospital 3333 Burnett Ave. Room D2.28 Cincinnati, OH 45229 DATE AND TIME 09/29/18 11:00am - 09/29/18 2:00pm This meeting will be hosted by Audrey Schering and Brandy Nicole Smith. Please let us know if you will be in attendance!
Patient Education and Support Meeting - Missouri
LOCATION Mound Ridge Retreat and Mission Center 31 Agape Lane Cook Station, MO 65449 DATE AND TIME 10/27/18 12:00pm - 10/27/18 2:00pm Question and Answer session by Porphyria Expert via Skype. Family and Friends are welcome to attend. Lunch will be provided.
Patient Education and Support Meeting - San Francisco
LOCATION UCSF Medical Center 505 Parnassus Ave. Room: S-157 San Francisco, CA 94143 DATE AND TIME 11/11/18 5:30pm - 11/11/18 7:00pm Seating is limited. Please RSVP Today!!
Contact Information
Is your contact information up to date? If not please give us a call @ 866-APF-3635 or Email to porphyrus@porphyriafoundation.com
Patient Education Meetings, PCT Study & Longitudinal Study
Friday - September 14, 2018 @ 06:30:11
Patient Education Meetings and more
Patient Education and Support Meeting LOCATION: Cincinnati Children's Hospital 3333 Burnett Ave. 2nd Floor, Section D Cincinnati, OH 45229 DATE AND TIME: 09/29/18 11:00am - 09/29/18 1:00pm Are you interested in hosting a Patient Education and Support Meeting? This is a valuable opportunity to bring patients, caregivers and supporters together to share common issues and to learn more about porphyria from an expert. The APF would like to help you coordinate your efforts. Please click on the link below, fill out the form attached and send it back to Edrin Williams via email at edrinw@porphyriafoundation.org or by mail to the APF office and someone will be in touch to map out the details of your meeting. We look forward to hearing from you!
Harvoni Study - PCT Do you have PCT? Are you interested in participating in research? Do you have Hepatitis C? If you answered YES to these questions, this is for you. We need YOU for a clinical trial! The purpose of this clinical trial is to assess whether Harvoni alone is an effective therapy in active PCT patients with Chronic Hepatitis C. Who can participate? Adult patients with PCT who also have Hepatitis C If you are interested in participating please contact Edrin Williams, Director of Patient Services at the APF office at 301.347.7166 for additional information.
Safe/Unsafe Drug Questionnaire for the Acute Porphyrias (AIP,VP,HCP & ADP) The APF is collaborating with researchers to identify new safe and unsafe drugs. We need your help. Are you experiencing adverse effects with any of your new medications? If so, please let us know in the questionnaire below how it affected you. We will share these results with our team of renowned Porphyria experts/researchers. They are in the process of updating our safe and unsafe drug list for the acute porphyrias. Your donations will help us educate physicians about the dangerous effects of unsafe drugs. Please click on the link below for the Questionnaire. Safe and Unsafe Drug List Questionnaire
Longitudinal Study Are you interested in participating in research? If so, we need YOU!! Our team of researchers are searching for people to participate in the Longitudinal Study. The purpose of this long-term follow-up study is to collect a large group of patients with the different types of porphyria and to provide a better understanding of the natural history of these disorders. The hope is that this information will help in developing new forms of treatment. Contact Edrin Williams, Director of Patient Services at the APF for additional information.
Kids With EPP Stories Shadow Jumpers Stories
Wednesday - September 12, 2018 @ 21:23:07
Kids With EPP Stories
INTERVIEWS HAPPENING NOW! We know growing up with EPP has its challenges. You may have to cover up and look different outside or may be stuck indoors on those hot sunny days but what's important is to know that you aren't alone out there. Many kids, just like you, are dealing with long days finding things to do to dodge the sun and ways to let loose at sun down. Here at Shadow Jumpers, we've developed a section for Kids to hear from people just like YOU! Check back here soon to read some amazing testimonial stories from kids in our EPP community. EPP may be hard, but it's something we continue to tackle together. Do you want to share your story for other EPP kids? Email us at shadowjumpers@porphyriafoundation.org or Amy@porphyriafoundation.org and we can provide you with the next steps!
Porphyria Post
Wednesday - September 12, 2018 @ 06:30:06
Hurricane Florence
Hurricane Florence is projected to be a strong Category 4 storm by the time it hits landfall sometime Thursday evening.
In preparation for the hurricane, please make sure that you are stocked up on non- perishable food items and bottled water.
To our members and doctors in the projected path, please stay safe and follow emergency management personnel orders.
Panhematin Study - Participants Needed!
Do you have AIP, VP or HCP? Are you interested in participating in research? If so, we are recruiting volunteers for the Panhematin Study.
Our researchers need people to volunteer for the Panhematin study. Completing this study will help prevent insurance companies from not paying for this treatment.
The purpose of this study is to determine if Panhematin is safe and effective for prevention of acute attacks of porphyria.
Please contact Edrin at the APF office (301.347.7166) if you are interested in participating.
Harvoni Study - PCT
Do you have PCT? Are you interested in participating in research? Do you have Hepatitis C? If you answered YES to these questions, this is for you.
We need YOU for a clinical trial!
The purpose of this clinical trial is to assess whether Harvoni alone is an effective therapy in active PCT patients with Chronic Hepatitis C.
Who can participate?
Adult patients with PCT who also have Hepatitis C
If you are interested in participating please contact Edrin Williams, Director of Patient Services at the APF office at 301.347.7166 for additional information.
Safe/Unsafe Drug Questionnaire for the Acute Porphyrias (AIP,VP,HCP & ADP - VERY IMPORTANT
The APF is collaborating with researchers to identify new safe and unsafe drugs. We need your help. Are you experiencing adverse effects with any of your new medications? If so, please let us know in the questionnaire below how it affected you. We will share these results with our team of renowned Porphyria experts/researchers. They are in the process of updating our safe and unsafe drug list for the acute porphyrias. Your donations will help us educate physicians about the dangerous effects of unsafe drugs. Please click on the link below for the Questionnaire.
Are you ready to be a medical hero? A new clinical trial with an oral investigational drug intended to reduce phototoxicity in people living with EPP has begun. Dont delay contact the APF to learn more at 1-866-APF-3635 or porphyrus@porphyriafoundation.org
"RememberResearch is the key to YOUR cure!"
Each Step Toward Finding an Effective Treatment is Important!
Patient Education and Support Meetings
Mark your calendars!
Saturday, September 29 Cincinnati Childrens Hospital 3333 Burnett Ave. Room S1.204 Cincinnati, OH 45229 11:00 AM 2:00 PM
Sunday, November 11 UCSF Medical Center 505 Parnassus Ave. Room: S-157 San Francisco, CA 94143 5:30 PM -7:00 PM
There are two main types of clinical studies: clinical trials and observational studies. In a clinical trial, there is some form of treatment intervention. There is no intervention in an observational study, which is aimed at observing patients to better understand the long-term course of their disease. Clinical trials are used to test new treatments before they are approved for use by the FDA. This type of trial gives patients a chance to try out a new medication in its early stages. As with any experiment, the result of a trial is not known before its conclusion. Your participation could help demonstrate a terrific treatment breakthrough, or it could help scientists discover that a new treatment does not work after all. There may be some risk involved from the treatment in a clinical trial. Participating in either a clinical trial or an observational study is a serious responsibility. Volunteering to participate could be a way to help yourself, affected family members and other patients by advancing medical and scientific knowledge of your condition. Some patients derive great satisfaction from assisting doctors in the study of their disease. Participation in a study can also mean a chance to meet a porphyria researcher in a clinical setting, and the consultation can be beneficial. For information on porphyria trials currently recruiting patients, call the APF office or visit NIHs clinical trials website: Clinical Trials of Medical Treatments: Why Volunteer? might also be useful reading as you think about whether youd like to participate or not.
Join the Porphyria Registry and LET THE GOVERNMENT KNOW THEY MUST PROVIDE FUNDING FOR PORPHYRIA RESEARCH!!!!! To join the Contact Registry, click here to open a page that lists all of the rare disease consortia. Scroll down the page until you come to the Porphyria Consortium and click on your type of porphyria. You will then be asked to complete a simple form including information about the date of your diagnosis, if you know it. If you have copies of your initial diagnostic lab results, you may want to have them handy when you go to the registry website. Porphyria experts have created this National Porphyria Registrya type of partnership between doctors and patients as a way for those with porphyria to share information about their health and treatment so physicians can learn from their experience and use that knowledge to enhance diagnosis, treatment and eventually find a cure for porphyria. It is the best means to prove that there are enough porphyria patients who want improved health care. If we don't speak up, we will be left behind when research funding is given. We DO NOT HAVE ENOUGH PEOPLE ON THE REGISTRY. Please join the registry. Joining the Porphyria Registry is anonymous and free of charge. All data will be stored in a secure, computerized database. No personal identifying information (such as your name, address, telephone number) will be given to anyone without your expressed approval. _________________________________________________ The registry is not linked to APF membership, but we hope you will join the American Porphyria Foundation too! So please consider joining the Contact Registry, and thank you for continuing to be a member of the APF. Doctors who study rare diseases see a relatively small number of sufferers over many years of practice. This Registry will give a big boost to medical and scientific understanding of porphyria by bringing together information from patients all over the country. If you need help enrolling in the registry contact our office toll free at 1-866-APF-3635.
https://www.rarediseasesnetwork.org/registry
Phase 2 clinical trial for MT-7117 EPP
Friday - September 7, 2018 @ 06:30:05
Phase 2 clinical trial for MT-7117 EPP
RESEARCH PARTICIPANTS NEEDED! Do you have Erythropoietic Protoporphyria (EPP)? We need you to participate in research! Get involved TODAY! Phase 2 clinical trial for MT-7117, an investigational drug for EPP, has begun. This trial is being conducted to assess effectiveness and safety of an oral investigational drug on sunlight exposure duration and tolerance in individuals with EPP. If you are interested in participating in this exciting new trial, contact the APF on 1.866.APF.3635 or porphyrus@porphyriafoundation.com. Subjects must have a confirmed diagnosis of EPP based on medical history and be between 18-70 years of age at screening. Participants must be willing to travel to the study site for all scheduled visits and be willing to cooperate and comply with the protocol restrictions and requirements. Exclusion criteria apply and may be discussed with the study site.
"RememberResearch is the key to YOUR cure!" Each Step Toward Finding an Effective Treatment is Important!
Erythropoietic Protoporphyria (EPP) and X-Linked Protoporphyria (XLP)
Wednesday - September 5, 2018 @ 06:30:07
Erythropoietic Protoporphyria (EPP) and X-Linked Protoporphyria (XLP)
Erythropoietic Protoporphyria is a disease of porphyrin metabolism characterized by abnormally elevated levels of protoporphyrin IX in erythocytes (mature blood cells), feces and plasma (the fluid portion of circulating blood), and by sensitivity to visible light. This sensitivity manifests itself as a burning sensation in the skin, followed by varying degrees of erythema (redness of the skin due to capillary dilation) and edema (swelling caused by excess fluids). Consequently, patients with the disorder commonly avoid exposure of the skin to strong light. They tend to choose indoor occupations and nocturnal work, or to venture outdoors while heavily clothed to protect the skin.
Diagnosis
EPP is diagnosed in patients with light sensitivity by testing blood and stool for the presence of abnormally high levels of protoporphyrin. Contrary to what is found in the other porphyrias, urine porphyrin levels remain within normal limits in EPP. When a smear of blood from a patient is examined under the fluorescence microscope, large numbers of red fluorescing erythrocytes are seen; these are not seen in persons who do not have this disorder. In addition, if the skin of the light-exposed areas of the body is examined under the light microscope, an amorphous homogeneous substance in and around the walls of small blood vessels of the upper papillary dermis will be seen. Histologic studies suggest that this substance is a neutral mucopolysaccharide, glycoprotein or mucoprotein. EPP is genetically transmitted as an autosomal recessive trait. Some relatives of patients may also have only slightly elevated levels of protoporphyrin but are asymptomatic, suggesting the existence of a carrier state.
Clinical Features
Most EPP patients experience the onset of photosensitivity before the age of six years and some as early as eighteen months. Patients report, in decreasing order of frequency, burning, swelling, itching and redness of the skin. After severe episodes of photosensitivity, some patients acquire shallow-depressed scars over the nose and cheeks and on the backs of hands. Some patients report only subjective symptoms of itching and burning and have no redness, swelling or scarring; these patients are often dismissed by their physicians as hypochondriacs, when in reality they have EPP. Thus, it is important for the physician to investigate for the presence of the disease in all patients who report itching and burning of the skin on exposure to light, even in the absence of objective findings. The amount of exposure to sun that a patient with EPP can tolerate varies from a few minutes to several hours. This photosensitivity is to light in the visible spectrum (400 to 700 nm). These wavelengths are not absorbed by window glass. Therefore, the symptoms can also develop from light passing through glass windows. About half of the patients report decreases in photosensitivity during winter. However, those engaging in skiing report that the light reflected by snow causes severe photosensitivity reactions. EPP is generally a benign disease. Many patients have somewhat decreased levels of hemoglobin and hematocrit (percentage of the volume of a blood sample occupied by cells). This finding usually requires no treatment. One reported case with severe hemolytic anemia (anemia caused by excessive destruction of red blood cells) improved after splenectomy (removal of the spleen). There also seems to be an increased frequency of cholelithiasis (presence of the formation of solid material in the gall bladder or bile duct), with several patients requiring cholecystectomy (removal of the gall bladder). Chemical analysis of the gallstones reveals high levels of protoporphyrin. To summarize, in EPP a decreased amount of the enzyme ferrochelatase leads to the accumulation of protoporphyrin in reticulocytes (young red blood cells that appear especially during regeneration of lost blood). This excess protoporphyrin leaks rapidly into the plasma from the maturing reticulocytes and young erythrocytes. The protoporphyrin is then partially cleared from the plasma by the liver and excreted into the bile (with or without some recirculation via the enterohepatic circulation). Accumulation of this protoporphyrin in the liver may lead, in rare cases, to serious liver disease.
Aides
Patients with EPP have found that topical sunscreens, which are effective in protecting against hypersensitivity to the sunburn spectrum of light, are ineffective as protective agents. Various systemic agents, such as antimalarials, inosine and vitamin E, have been tried but with little success. Patients with EPP may develop liver abnormalities due to an excess deposition of protoporphyrin in that organ. Hence, drugs that can impair bile flow, cholestasis, as well as estrogens, should be given cautiously. Cholestyramine ingestion may lower porphyrin levels in some patients. Some patients with EPP report that drinking alcoholic beverages increases their photosensitivity. It is probably wise for all EPP patients to avoid or greatly reduce alcohol consumption. Individuals with EPP may also need to wear protective clothing, such as garments with long sleeves and trousers: each one has to decide the amount of this sort of protection needed. Certain kinds of fabric, such as denim, are quite light protective. Also certain plastic materials, such as Scotchtint, filters out harmful rays and can be used on home and automobile windows.
Lauren Fabean Erythropoietic Protoporphyria (EPP)
Wednesday - August 29, 2018 @ 15:56:49
Lauren Fabean
Erythropoietic Protoporphyria (EPP)
My name is Lauren Fabean, and I am a sun-hater! I enjoy rainy days and feel an immediate sense of relief when I see "considerable cloudiness" in a weather forecast. I was diagnosed with EPP at the age of 12. After enduring the worst reaction of my life while participating in a weekend softball tournament (I really hated playing softball, too!), my mom started seeking answers to what was happening to me and why, and the journey to my diagnosis began. It took me a long time to get used to having EPP, and to learn what preventative measures worked for me. I also had to learn how much sunlight I could handle before a reaction set in. This was really hard. 23 years later, I still have days when I think I will be ok in a bit of sunlight, and the next thing I know I'm kicking myself for allowing a reaction to occur. This is what happens to me when I am exposed to too much sunlight for too long of a time period: I feel an intense burning sensation in my hands, feet, and/or face. When trying to cool the areas, I feel an extreme tingling sensation if I am applying something that is too cold. I need to find a happy medium. My skin is also very sensitive during this time; a simple bump or scrape against an object leaves a lasting, uncomfortable tingling sensation. A bad reaction can take days to go away. In the summer, even if I'm very careful, I have come to terms with the fact that I will always be living with a slight reaction here or there. Unless I want to just hide inside every day. What works for me: I am a firm believer in sunscreen, not just because of EPP, but because the sun is so damaging to our skin and it's so important to take care of it. I have spent a lot of money on good clothing with SPF and hats, and I can be seen carrying an umbrella (my "sunbrella" as I call it) at times. The best thing for me to do is simply stay away from the rays. I struggled a bit growing up, and did feel lonely at times. However, the worst part of having EPP in my adult life is the fact that I cannot play or swim with my daughter in the summer. I get to sit on the sidelines, hiding in my shade, while I watch my husband enjoy all of those things that I wish I could do. This disease has taught me a lot. Most importantly, I realize that I am very lucky. There are many others who suffer far worse than I do when exposed to sunlight. EPP is mostly a huge inconvenience in my life, but its something that Ive accepted. So, if you see me running desperately to the shade or hiding under my hats and umbrella, dont feel bad for me or think Im weird. Thats just me living with my sun thing.
Porphyria Post
Friday - August 24, 2018 @ 07:00:09
Holiday Office Closure
The American Porphyria Foundation will be closed on Monday, September 3, 2018 and will re-open for normal business hours on Tuesday, September 4, 2018 in observance of Labor Day. Enjoy your weekend!
Take a look at our reminders below!
Safe/Unsafe Drug Questionnaire for the Acute Porphyrias (AIP,VP,HCP & ADP - VERY IMPORTANT
The APF is collaborating with researchers to identify new safe and unsafe drugs. We need your help. Are you experiencing adverse effects with any of your new medications? If so, please let us know in the questionnaire below how it affected you. We will share these results with our team of renowned Porphyria experts/researchers. They are in the process of updating our safe and unsafe drug list for the acute porphyrias. Your donations will help us educate physicians about the dangerous effects of unsafe drugs. Please click on the link below for the Questionnaire.
Do you have AIP, VP or HCP? Are you interested in participating in research? If so, we are recruiting volunteers for the Panhematin Study.
Our researchers need people to volunteer for the Panhematin study. Completing this study will help prevent insurance companies from not paying for this treatment.
The purpose of this study is to determine if Panhematin is safe and effective for prevention of acute attacks of porphyria.
Please contact Edrin at the APF office (301.347.7166) if you are interested in participating.
Harvoni Study - PCT
Do you have PCT? Are you interested in participating in research? Do you have Hepatitis C? If you answered YES to these questions, this is for you.
We need YOU for a clinical trial!
The purpose of this clinical trial is to assess whether Harvoni alone is an effective therapy in active PCT patients with Chronic Hepatitis C.
Who can participate?
â?¢ Adult patients with PCT who also have Hepatitis C
If you are interested in participating please contact Edrin Williams, Director of Patient Services at the APF office at 301.347.7166 for additional information.
EPP Clinical Trials Participants Needed!
Are you ready to be a medical hero? A new clinical trial with an oral investigational drug intended to reduce phototoxicity in people living with EPP has begun. Dont delay contact the APF to learn more at 1-866-APF-3635 or porphyrus@porphyriafoundation.org
"RememberResearch is the key to YOUR cure!"
Each Step Toward Finding an Effective Treatment is Important!
This meeting will be hosted by Audrey Schering and Brandy Nicole Smith. Please let us know if you will be in attendance!
I'll be there!
Maybe
I can't make it
FDA- Chronic Pain PFDD Meeting docket comments
The last day to send in docket comments for the Chronic Pain Patient-Focused Drug Development (PFDD) Meeting is Monday, September 10, 2018! We encourage patients with chronic pain and other stakeholders to submit written comments through the regulations.gov website: https://www.regulations.gov/document?D=FDA-2018-N-1621-0001. We carefully review and consider all comments.
In addition, we want to let you all know that the meeting recording, materials, and full transcript are available on fda.gov: https://www.fda.gov/Drugs/NewsEvents/ucm603093.htm.
Thank you for participating in this meeting and in sharing your experience. If you have any questions, please email: Patient.Focused@fda.hhs.gov.
Acute Intermittent Porphyria: Symptoms, Triggers, and Other Factors Common symptoms of acute intermittent porphyria (AIP)
Wednesday - August 22, 2018 @ 21:30:27
Acute Intermittent Porphyria: Symptoms, Triggers, and Other Factors Common symptoms of acute intermittent porphyria (AIP) AIP can cause many different symptoms that tend to come and go. When someone with AIP has symptoms, the episode is called an AIP attack. A person may have certain symptoms during one attack but different symptoms during other attack. Severe abdominal pain â?¢ Severe abdominal pain is the most common symptom of AIP. More than 85% of people have abdominal pain during AIP attacks. â?¢ The pain often lasts for hours or days. â?¢ The pain tends to begin slowly and become worse. â?¢ The pain tends to be all over and not in one small area. â?¢ The pain may start in the chest or back and move to the abdomen. â?¢ Pain medicines such as Advil (ibuprofen) or Tylenol (acetaminophen) usually dont help much. This is because the pain is caused by nerves. Nausea or vomiting â?¢ Nausea or vomiting is common during AIP attacks. â?¢ People with AIP often have nausea or vomiting along with abdominal pain. Constipation â?¢ Constipation is common during AIP attacks. â?¢ Loss of bladder control may go along with constipation. Dark or reddish urine â?¢ Urine that turns reddish or dark over time when exposed to light and air is common during AIP attacks. â?¢ The change in urine color is due to certain chemicals in the urine during AIP attacks. Muscle weakness â?¢ Muscle weakness is common during AIP attacks. â?¢ Muscle weakness usually begins in the arms and often in the shoulders. 2 Common symptoms of AIP (cont.) Pain in the arms, legs, back, chest, neck, or head â?¢ Abdominal pain is the most common symptom of AIP. But pain in other areas is also common. â?¢ Pain may start in the chest or back and move to the abdomen. â?¢ Pain medicines such as Advil (ibuprofen) or Tylenol (acetaminophen) usually dont help much. This is because the pain is caused by nerves. Fast heartbeat â?¢ Fast heartbeat is common during AIP attacks. High blood pressure â?¢ High blood pressure is common during AIP attacks. Emotional changes â?¢ People with AIP often experience emotional changes. â?¢ Certain emotional changes may mean that an attack is coming on. These changes include: Feeling anxious or restless o Having a hard time sleeping â?¢ Over time, more severe emotional changes may develop, such as: o Feeling upset or confused o Seeing or hearing things that arent there Feeling sad or depressed Frequency of symptoms â?¢ AIP attacks are acute and intermittent, meaning they tend to come and go. Gender and AIP â?¢ AIP attacks are 4 to 5 times more common in women than in men. â?¢ AIP attacks are more common during child-bearing years, often in women in their thirties. 3 Possible triggers for AIP symptoms If you have AIP, certain things may upset your bodys chemical balance enough to cause symptoms. One or a combination of triggers can lead to an AIP attack. Monthly cycle/hormone changes â?¢ AIP attacks are more common during the two weeks before periods start. â?¢ Hormones fluctuate the most during these two weeks. Monthly cycle/hormone changes â?¢ Dietary changes, such as going on a diet or fasting, are common triggers of AIP attacks. Medicines â?¢ Taking certain kinds of medicines, including estrogens (oral contraceptives), barbiturates (sometimes used to aid sleep or treat epilepsy), steroids (body hormone-like drugs), and some antibiotics, can trigger AIP attacks. â?¢ Starting a new medicine may trigger an AIP attack. Substance use â?¢ Drinking alcohol, smoking cigarettes, or using illegal drugs such as amphetamines or cocaine may trigger AIP attacks. Sickness or infections â?¢ Stress on the body caused by infections, illness, or surgery may trigger AIP attacks. Family history of AIP â?¢ AIP is caused by a partial lack of an enzyme that the body needs. AIP is inheritedpeople with the enzyme deficiency are born with it. â?¢ You can inherit the enzyme deficiency from one parent. If you do, you can have AIP. But most people with the enzyme deficiency never have symptoms. Untreated AIP attacks can cause serious, long-term health problems. If you think you may have AIP, work closely with your doctor to get a correct diagnosis and treatment that is right for you. 4 References Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, Desnick RJ. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142:439-450. Besur S, Hou W, Schmeltzer P, Bonkovsky HL. Clinically important features of porphyrin and heme metabolism and the porphyrias. Metabolites. 2014;4:977-1006. Crimlisk HL. The little imitatorporphyria: a neuropsychiatric disorder. J Neurol Neurosurg Psychiatry. 1997;62:319-328. Elder GH, Hift RJ. Treatment of acute porphyria. Hosp Med. 2001;62(7):422-425. Gonzalez-Arriaza HL, Bostwick JM. Acute porphyrias: a case report and review. Am J Psych. 2003;160(3);450-458. Pischik E, Kauppinen R. An update of clinical management of acute intermittent porphyria. Appl Clin Gen. 2015;8:201-214. Ventura P, Cappellini MD, Rocchi E. The acute porphyrias: a diagnostic and therapeutic challenge in internal and emergency medicine. Intern Emerg Med. 2009;4:297-308.
Porphyria Post
Thursday - August 9, 2018 @ 06:30:11
Porphyria Post
Harvoni Study - PCT
Do you have PCT? Are you interested in participating in research? Do you have Hepatitis C? If you answered YES to these questions, this is for you.
We need YOU for a clinical trial!
The purpose of this clinical trial is to assess whether Harvoni alone is an effective therapy in active PCT patients with Chronic Hepatitis C.
Who can participate?
â?¢ Adult patients with PCT who also have Hepatitis C
If you are interested in participating please contact Edrin Williams, Director of Patient Services at the APF office at 301.347.7166 for additional information.
EPP Clinical Trials Participants Needed!
Are you ready to be a medical hero? A new clinical trial with an oral investigational drug intended to reduce phototoxicity in people living with EPP has begun. Dont delay contact the APF to learn more at 1-866-APF-3635 or porphyrus@porphyriafoundation.org
"RememberResearch is the key to YOUR cure!"
Each Step Toward Finding an Effective Treatment is Important!
Safe/Unsafe Drug Questionnaire for the Acute Porphyrias (AIP,VP,HCP & ADP
The APF is collaborating with researchers to identify new safe and unsafe drugs. We need your help. Are you experiencing adverse effects with any of your new medications? If so, please let us know in the questionnaire below how it affected you. We will share these results with our team of renowned Porphyria experts/researchers. They are in the process of updating our safe and unsafe drug list for the acute porphyrias. Your donations will help us educate physicians about the dangerous effects of unsafe drugs. Please click on the link below for the Questionnaire.
The APF is fighting hard for Scenesse to be granted Priority Review status at the FDA. This is the fastest possible review process for a new treatment through the FDA. Scenesse has proven to be an effective treatment with nearly 6,700 worldwide SAFE doses implanted for the treatment of EPP. There IS no current FDA-approved treatment available for this extremely painful phototoxic disease. As a TEAM we are much more effective. Please use this Facebook frame to show your support for our cause and to help gain attention to the much needed Priority Review and approval of the EPP treatment Scenesse!
Do you have PCT? Are you interested in participating in research? Do you have Hepatitis C? If you answered YES to these questions, this is for you. We need YOU for a clinical trial!
The purpose of this clinical trial is to assess whether Harvoni alone is an effective therapy in active PCT patients with Chronic Hepatitis C.
Who can participate? Adult patients with PCT who also have Hepatitis C
If you are interested in participating please contact Edrin Williams, Director of Patient Services at the APF office at 301.347.7166 for additional information.
EPP Clinical Trials Participants Needed!
Please contact the APF on 866-APF-3635 to request information on these new clinical trials. There are six study sites across the US that need your participation. Do you want a new treatment for EPP? then come be part of the solution. There are 4 Sites that are currently accepting patients: Mount Sinai - New York City, NY Wake Forest Baptist - Winston Salem, NC University of Miami - Miami, FL University of Texas Medical Branch - Galveston, TX Sites to open soon: University of California San Francisco - San Francisco, CA University of Utah - Salt Lake City, UT We need your participation! Contact us today!
Rememberâ?¦Research is the Key to Your Cure!! Every step along the way is important.
Light the Moment 2018
The Stuhlsatz Family was the recipient of Light the Moment 2018. We are excited to share with you that they are headed to Disney World this week! The Shadow Jumpers team worked very hard to make sure that they have an experience of a lifetime. Stay tuned for more once they return from this exciting trip.
Safe/Unsafe Drug Questionnaire for the Acute Porphyrias (AIP,VP,HCP & ADP)
The APF is collaborating with researchers to identify new safe and unsafe drugs. We need your help. Are you experiencing adverse effects with any of your new medications? If so, please let us know in the questionnaire below how it affected you. We will share these results with our team of renowned Porphyria experts/researchers. They are in the process of updating our safe and unsafe drug list for the acute porphyrias. Your donations will help us educate physicians about the dangerous effects of unsafe drugs. Please click on the link below for the Questionnaire.
Pub MD specials on each type of Porphyria Click on the link
Balwani M, Wang B, Anderson KE, Bloomer JR, Bissell DM, Bonkovsky HL, Phillips JD, Desnick RJ;PorphyriasConsortium of the Rare Diseases Clinical Research Network.
Do you have AIP, VP or HCP? Are you interested in participating in research? If so, we are recruiting volunteers for the Panhematin Study.
Our researchers need people to volunteer for the Panhematin study. Completing this study will help prevent insurance companies from not paying for this treatment.
The purpose of this study is to determine if Panhematin is safe and effective for prevention of acute attacks of porphyria.
Please contact Edrin at the APF office (301.347.7166) if you are interested in participating.
EPP on Fox News - Thursday, August 2nd at 10pm!
Camp Sundown 2018 was extra special for our porphyria community - the first week was dedicated to children with EPP. Fox News caught wind of this special event and came to see what camp and living with photosensitivity is all about. Tune in tomorrow night - August 2nd - at 10pm to see the segment. The APF will have access after it airs and we will be sure to share with all of you.
Hearing Hoofbeats, Thinking Zebras
Click on the link below to view a story by our patient member Eryn Sallee. Thank you, Eryn, for sharing your story!
Please contact the APF on 866-APF-3635 to request information on these new clinical trials. There are seven study sites across the US that need your participation. Do you want a new treatment for EPP? then come be part of the solution.
Rememberâ?¦Research is the Key to Your Cure!! Every step along the way is important.
Harvoni Study - PCT
Do you have PCT? Are you interested in participating in research? Do you have Hepatitis C? If you answered YES to these questions, this is for you.
We need YOU for a clinical trial!
The purpose of this clinical trial is to assess whether Harvoni alone is an effective therapy in active PCT patients with Chronic Hepatitis C.
Who can participate?
·Adult patients with PCT who also have Hepatitis C
If you are interested in participating please contact Edrin Williams, Director of Patient Services at the APF office at 301.347.7166 for additional information.
Some types of porphyria are inherited in an autosomal dominant pattern, which means one copy of the gene in each cell is mutated. This single mutation is sufficient to reduce the activity of an enzyme needed for heme production, which increases the risk of developing signs and symptoms of porphyria. Autosomal dominant porphyrias include acute intermittent porphyria, most cases of erythropoietic protoporphyria, hereditary coproporphyria, and variegate porphyria. Although the gene mutations associated with some cases of porphyria cutanea tarda also have an autosomal dominant inheritance pattern, most people with this form of porphyria do not have an inherited gene mutation.
Other porphyrias are inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but do not show signs and symptoms of the condition. Porphyrias with an autosomal recessive pattern of inheritance include ALAD deficiency porphyria, congenital erythropoietic porphyria, and some cases of erythropoietic protoporphyria.
When erythropoietic protoporphyria is caused by mutations in the ALAS2 gene, it has an X-linked dominant pattern of inheritance. The ALAS2 gene is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the gene in each cell may be sufficient to cause the disorder. In males (who have only one X chromosome), a mutation in the only copy of the gene in each cell causes the disorder. Males may experience more severe symptoms of the disorder than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Mutations in the UROD gene are related to both porphyria cutanea tarda and hepatoerythropoietic porphyria. Individuals who inherit one altered copy of the UROD gene are at increased risk for porphyria cutanea tarda. (Multiple genetic and nongenetic factors contribute to this condition.) People who inherit two altered copies of the UROD gene in each cell develop hepatoerythropoietic porphyria.
The UROS gene provides instructions for making an enzyme known as uroporphyrinogen III synthase. This enzyme is involved in the production of a molecule called heme. Heme is vital for all of the body's organs, although it is most abundant in the blood, bone marrow, and liver. Heme is an essential component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood).
The production of heme is a multi-step process that requires eight different enzymes. Uroporphyrinogen III synthase is responsible for the fourth step in this process, in which hydroxymethylbilane (the product of the third step) is rearranged to form uroporphyrinogen III. In subsequent steps, four other enzymes produce and modify compounds that ultimately lead to heme.
Historic New Drug Application for the use of SCENESSE in rare metabolic disorder EPP
Friday - July 27, 2018 @ 07:00:02
Monday, 25 June 2018 10:00
Historic New Drug Application for the use of SCENESSE in rare metabolic disorder EPP
EXECUTIVE SUMMARY
First NDA for SCENESSE (afamelanotide 16mg) in the United States
First-line therapy proposed as systemic photoprotection for EPP patients
Submission of data from five clinical trials in EPP, pooled data, Compassionate Use, Special Access Schemes, and real-world experience from European use
Nearly 6,700 afamelanotide doses administered to more than 800 patients
Priority Review requested to FDA
CLINUVEL PHARMACEUTICALS LTD (ASX: CUV; XETRA-DAX: UR9; NASDAQ INTERNATIONAL DESIGNATION: CLVLY) today announced that it has completed the submission of a New Drug Application (NDA) for its drug SCENESSE (afamelanotide 16 mg) as the first proposed therapy for patients with the rare metabolic disorder erythropoietic protoporphyria (EPP) in the United States. An approved NDA will allow CLINUVEL to make SCENESSE available to adult EPP patients in the US as a first-line therapy.
REGULATORY TIMELINESSince 2005, CLINUVEL has been in regular and frequent communication with the US Food and Drug Administration (FDA) to discuss the development program for SCENESSE as a preventative treatment for adult EPP patients.
The FDA granted SCENESSEorphan drug designation for EPP in 2008. In July 2016 the FDA awarded Fast Track Designation allowing the Company to make its final regulatory submission.
On 24 October 2016, the FDA initiated a public Scientific Workshop on EPP in Silver Spring, Maryland, to gain more, and first-hand, information on the characteristics and impact of the disease from EPP patients and their families.
In November 2016, the FDA concluded that CLINUVELs scientific data were ready to be filed as part of a rolling review for evaluation under an NDA. The rolling review enabled the Company to make its NDA submission in parts; the FDAs scientific review time starts once the final module has been filed and validated. The regulatory validation of the submitted dossier is now expected to take two months, after which a target date for the completion of the review may be provided (PDUFA date).
CLINUVEL has filed for Priority Review and awaits the FDAs answer to the request for scientific review on an abbreviated basis. Under a Priority Review, the FDA aims to arrive at a benefit versus risk assessment within six months from final submission, whereas a standard scientific review is estimated by the FDA to take up to ten months.
In October 2014 the European Medicines Agency (EMA) granted SCENESSE marketing authorisation.1 The product was launched in Europe in June 2016.
--
Estimated FDA Timeline
22 June 2018 NDA Submission SCENESSE
22 August 2018 Validation NDA
2019 Approval or Complete Response*
* Scientific review time depends on review status: Standard Review or Priority Review --
NDA DOSSIER FOR SCENESSEIN THE TREATMENT OF EPP
As part of the NDA dossier, CLINUVEL submitted data and analyses from five clinical trials in EPP, data from Compassionate Use and Special Access Schemes, and data from the real-world experience of EPP patients receiving treatment in Europe. The data set consists of nearly 6,700 doses in more than 800 patients.
The safety profile of SCENESSEhas been positive to date and includes longer term exposure (over 12 years) from patients involved in multiple consecutive programs in Europe. A post-authorisation pharmacovigilance plan to monitor patients in the US long-term is part of the NDA submission.
COMMENTARY
CLINUVEL has arrived at an historic moment, CLINUVELs Chief Scientific Officer, Dr Dennis Wright said. The scientific community started publishing on the use of melanocortins in man in 1987. It is owing to CLINUVELs scientific team that new boundaries were broken to arrive at an innovative product providing systemic photoprotection in a genetic disorder for which there was no treatment. Todays NDA filing is the direct consequence of the many decisions our team has taken to persevere in the introduction of SCENESSE, which facilitates a life for EPP patients that they had never known before. The time lapsed since first scientific concept not only illustrates the complexity of launching a novel molecule, but also shows the concentration of resources and passion necessary to realise this dream for patients worldwide.
We consciously awaited validation of the EMAs Annual Report to add real world data to our FDA submission, since it is apparent that real-time data from patients demonstrate ultimately whether a novel pharmaceutical product offers the expected benefit or not. Since introducing SCENESSEin Europe we have seen that over 98% of the EPP patients on treatment request the drug for a second treatment year. I specifically wanted to see the real clinical demand and, indirectly, effectiveness of the therapy, incorporated into our final NDA submission, Dr Wright said.
In my capacity as Chair I have overseen the testing decisions our management team faced to implement a considered and well-thought out development program one which I and many experts in the field view as one of the most remarkable turnarounds in pharmaceuticals in the Southern Hemisphere, CLINUVELs Chair, Stan McLiesh said. Vision, planning, diligence and patience was borne by our teams and led to todays NDA filing. I congratulate Dr Wolgen and Dr Wright, the CLINUVEL staff and management team for their persistence and delivery. We now keenly await the FDAs validation and review of the NDA submission.
End
1SCENESSE (afamelanotide 16mg) is approved in Europe as an orphan medicinal product for the prevention of phototoxicity in adult patients with EPP. Information on the product can be found on CLINUVELs website at www.clinuvel.com.
About CLINUVEL PHARMACEUTICALS LIMITED
CLINUVEL PHARMACEUTICALS LTD (ASX: CUV; NASDAQ INTERNATIONAL DESIGNATION ADR: CLVLY; XETRA-DAX: UR9) is a global biopharmaceutical company focused on developing and delivering treatments for patients with a range of severe genetic and skin disorders. As pioneers in photomedicine and understanding the interaction of light and human biology, CLINUVELs research and development has led to innovative treatments for patient populations with a clinical need for photoprotection and repigmentation. These patient groups range in size from 5,000 to 45 million worldwide. CLINUVELs lead compound, SCENESSE(afamelanotide 16mg), was approved by the European Commission in 2014 for the prevention of phototoxicity (anaphylactoid reactions and burns) in adult patients with erythropoietic protoporphyria (EPP). More information on EPP can be found at http://www.epp.care.
Headquartered in Melbourne, Australia, CLINUVEL has operations in Europe, Switzerland, the US and Singapore.
SCENESSE is a registered trademark of CLINUVEL PHARMACEUTICALS LTD.
About erythropoietic protoporphyria (EPP)
Erythropoietic protoporphyria (EPP) is a rare metabolic disorder that causes severe anaphylactoid reactions to light (phototoxicity). Patients incur physical burns and ulcers, and are in state of crisis following light exposure, summarised as phototoxicity. This usually occurs within minutes of exposure to bright lights, especially sunlight.
EPP symptoms can be acute, or delayed (subacute), most often expressed as generalised oedema, effusion in tissue and distortion of the skin. As little as a few minutes of light outdoors (even when it is overcast or transmitted through a window) or artificial light exposure may be sufficient to evoke EPP symptoms.
Phototoxicity is unresponsive to traditional pain and burn management techniques and patients can be incapacitated for days before reactions subside. Most patients withdraw from light exposure in order to manage their phototoxic symptoms.
This release to the Australian Securities Exchange and to press may contain forward-looking statements, including statements regarding future results, performance or achievements. These statements involve known and unknown risks, uncertainties and other factors which may cause CLINUVELs actual results, performance or achievements to be materially different from any future results, performances or achievements expressed or implied by the forward-looking statements. These statements reflect our current views with respect to future events and are based on assumptions and subject to risks and uncertainties. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Some of the factors that could affect the forward-looking statements contained herein include: that the FDA may require additional studies beyond the studies planned for product candidates or may not provide regulatory clearances, including for SCENESSE; that the FDA may not provide regulatory approval for any use of SCENESSE or that the approval may be limited; that CLINUVEL may never file an NDA for SCENESSEregulatory approval in the US; that the Company may not be able to access adequate capital to advance its vitiligo programs; that the Company may not be able to retain its current pharmaceutical and biotechnology key personnel and knowhow for further development of its product candidates or may not reach favourable agreements with potential pricing and reimbursement agencies in Europe and the US.
Level 6, 15 Queen Street T +61 3 9660 4900 www.clinuvel.com
The PPOX gene provides instructions for making an enzyme known as protoporphyrinogen oxidase. This enzyme is involved in the production of a molecule called heme. Heme is vital for all of the body's organs, although it is most abundant in the blood, bone marrow, and liver. Heme is an essential component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood).
The production of heme is a multi-step process that requires eight different enzymes. Protoporphyrinogen oxidase is responsible for the seventh step in this process, in which two hydrogen atoms are removed from protoporphyrinogen IX (the product of the sixth step) to form protoporphyrin IX. In the final step, another enzyme modifies protoporphyrin IX by inserting an iron atom to produce heme.
The UROD gene provides instructions for making an enzyme known as uroporphyrinogen decarboxylase. This enzyme is involved in the production of a molecule called heme. Heme is vital for all of the body's organs, although it is most abundant in the blood, bone marrow, and liver. Heme is an essential component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood).
The production of heme is a multi-step process that requires eight different enzymes. Uroporphyrinogen decarboxylase is responsible for the fifth step in this process, in which carbon and oxygen atoms are removed from uroporphyrinogen III (the product of the fourth step) to form coproporphyrinogen III. In subsequent steps, three other enzymes produce and modify compounds that ultimately lead to heme.
Porphyria Post
Wednesday - July 25, 2018 @ 07:00:14
Harvoni Study - PCT
Do you have PCT? Are you interested in participating in research? Do you have Hepatitis C? If you answered YES to these questions, this is for you.
We need YOU for a clinical trial!
The purpose of this clinical trial is to assess whether Harvoni alone is an effective therapy in active PCT patients with Chronic Hepatitis C.
Who can participate?
·Adult patients with PCT who also have Hepatitis C
If you are interested in participating please contact Edrin Williams, Director of Patient Services at the APF office at 301.347.7166 for additional information.
PRIORITY REVIEW letters sent to the FDA!!
You are an amazing community! We had a great response to the call urgent call to action for patient letters.511 letters, a petition, and many poignant photos were sent to the desks of decision-makers at the FDA requesting Priority Review for SCENESSE (afamelanotide 16mg).
The new drug application for this treatment was submitted to the FDA on June 22.The FDA has sixty days to decide if the filing will receive Priority Review (6 months) or Standard Review (10 months).Our EPP patients have waiting long enough for treatment.We hope receipt of these letters will help urge the FDA to select the quicker path to treatment.Nearly 6,700 doses have been implanted in over 800 patients.
WANT TO DO MORE?Check out these In-District Lobby Days below and call your legislators to ask them for help.
You can participate in In-District Lobby Days!
From July 31st through September 4th, rare disease advocates from across the country will surge into the district offices of their Members of Congress to advocate for legislation benefiting the rare disease community.
The registration deadline for this event has passed. However, you can still contact your legislators offices while they are in-district.
The American Porphyria Foundation can help you with issues and talking points important to each type of porphyria.
Please contact the APF on 866-APF-3635 to request information on these new clinical trials.There are seven study sites across the US that need your participation. Do you want a new treatment for EPP? then come be part of the solution.
Rememberâ?¦Research is the Key to Your Cure!! Every step along the way is important.
HFE gene homeostatic iron regulator & HMBS gene hydroxymethylbilane synthase
Monday - July 23, 2018 @ 07:00:12
The HFE gene provides instructions for producing a protein that is located on the surface of cells, primarily liver and intestinal cells. The HFE protein is also found on some immune system cells.
The HFE protein interacts with other proteins on the cell surface to detect the amount of iron in the body. The HFE protein regulates the production of another protein called hepcidin, which is considered the "master" iron regulatory hormone. Hepcidin is produced by the liver, and it determines how much iron is absorbed from the diet and released from storage sites in the body. When the proteins involved in iron sensing and absorption are functioning properly, iron absorption is tightly regulated. On average, the body absorbs about 10 percent of the iron obtained from the diet.
The HFE protein also interacts with two proteins called transferrin receptors; however, the role of these interactions in iron regulation is unclear.
The HMBS gene provides instructions for making an enzyme known as hydroxymethylbilane synthase. This enzyme is involved in the production of a molecule called heme. Heme is vital for all of the body's organs, although it is most abundant in the blood, bone marrow, and liver. Heme is an essential component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood).
The production of heme is a multi-step process that requires eight different enzymes. Hydroxymethylbilane synthase is responsible for the third step in this process, which combines four molecules of porphobilinogen (the product of the second step) to form a compound called hydroxymethylbilane. In subsequent steps, five other enzymes produce and modify compounds that ultimately lead to heme.
The CPOX gene provides instructions for making an enzyme known as coproporphyrinogen oxidase. This enzyme is involved in the production of a molecule called heme. Heme is vital for all of the body's organs, although it is most abundant in the blood, bone marrow, and liver. Heme is an essential component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood).
The production of heme is a multi-step process that requires eight different enzymes. Coproporphyrinogen oxidase is responsible for the sixth step in this process, the removal of carbon and oxygen atoms from coproporphyrinogen III (the product of the fifth step) to form protoporphyrinogen IX. In subsequent steps, two other enzymes modify protoporphyrinogen IX and incorporate an iron atom to produce heme.
The FECH gene provides instructions for making an enzyme known as ferrochelatase. This enzyme is involved in the production of a molecule called heme. Heme is vital for all of the body's organs, although it is most abundant in the blood, bone marrow, and liver. Heme is an essential component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood).
The production of heme is a multi-step process that requires eight different enzymes. Ferrochelatase is responsible for the eighth and final step in this process, in which an iron atom is inserted into the center of protoporphyrin IX (the product of the seventh step) to form heme.
Porphyria Post July 20th, 2018
Friday - July 20, 2018 @ 07:00:08
Porphyria Post
Last Call - Request Priority Review of Scenesse
URGENT NEED FOR LETTERS
The new drug application for Scenesse has been submitted to the FDA - and it is time to advocate! We need your letters to the FDA Commisioner requesting Priority Review. Please send your letter immediately to add to our growing pile!! It is starting to look impressive!
All letters must be signed and emailed to the APF this weekend. We will submit the letters to the FDA on Monday July 23, 2018.
We will present all of the letters at once to the FDA! Right now we have 478 letters. Let's get to 500! Please send letters today! Time is of the Essence!
We have moved.
The American Porphyria Foundation is excited to announce out move to Bethesda, Maryland - next door to our nation's capital. Please update your records with our new address.
You can now support the APF through the AmazonSmile program! Amazon will donate 0.5-0.8% of the price of your eligible purchases to the APF, at no cost to you. Please make the APF your choice of a charitable organization. Support porphyria research while shopping!
Note, this program to provide donations to the APF will ONLY be available to shoppers who visit Amazon via a special web address, namely, www.smile.amazon.com instead of the normal www.amazon.com homepage.
It is easy and free! AmazonSmile is the same Amazon you know same products, same prices, same service.
Thank you for supporting us! Please follow the link:
The new drug application for Scenesse has been submitted to the FDA - and it is time to advocate! We need your letters to the FDA Commisioner requesting Priority Review. Please send your letter immediately to add to our growing pile!! It is starting to look impressive!
All letters must be signed and emailed to the APF this weekend. We will submit the letters to the FDA on Monday July 23, 2018.
We will present all of the letters at once to the FDA! Right now we have 478 letters. Let's get to 500! Please send letters today! Time is of the Essence!
Clinical Trial for EPP!
Research is the key to your cure! What is the most important element in research? YOU! You now have an opportunity to participate in a trial for a new EPP investigational drug. Contact the APF for details. YOU are our hope for the future. Contact the APF on 1-866-APF-3635 or email porphyrus@porphyriafoundation.com.
An oral investigational drug has been developed with the potential to increase sunlight duration and tolerance in individuals with EPP. This phase II clinical trial is needed to understand if the investigational drug works and to ensure that it is safe. The study duration is 24 weeks including follow-up. You must be age 18-70 to participate
and have a confirmed diagnosis of EPP. All travel is included and will be arranged by a concierge service.
"RememberResearch is the key to YOUR cure!
Each Step Toward Finding an Effective Treatment is Important!
Harvoni Study - PCT
The purpose of this clinical trial is to assess whether Harvoni alone is an effective therapy in active PCT patients with Chronic Hepatitis C.
Who can participate?
·Adult patients with PCT who also have Hepatitis C
If you are interested in participating please contact Edrin Williams, Director of Patient Services at the APF office at 301.347.7166 for additional information.
ALAD Gene aminolevulinate dehydratase & ALAS2 gene 5'-aminolevulinate synthase 2
Wednesday - July 18, 2018 @ 07:00:10
Learn about each gene-
aminolevulinate dehydratase
TheALADgene provides instructions for making an enzyme known as delta-aminolevulinate dehydratase. This enzyme is involved in the production of a molecule called heme. Heme is vital for all of the body's organs, although it is found mostly in the blood, bone marrow, and liver. Heme is an essential component of several iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood).
The production of heme is a multi-step process that requires eight different enzymes. Delta-aminolevulinate dehydratase is responsible for the second step in this process, which combines two molecules of delta-aminolevulinic acid (the product of the first step) to form a compound called porphobilinogen. In subsequent steps, four molecules of porphobilinogen are combined and then modified to produce heme.
ALAS2 gene
5'-aminolevulinate synthase 2
The ALAS2 gene provides instructions for making an enzyme called 5'-aminolevulinate synthase 2 or erythroid ALA-synthase. This version of the enzyme is found only in developing red blood cells called erythroblasts.
ALA-synthase plays an important role in the production of heme. Heme is a component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood). Heme is vital for all of the body's organs, although it is most abundant in the blood, bone marrow, and liver.
The production of heme is a multi-step process that requires eight different enzymes. ALA-synthase is responsible for the first step in this process, the formation of a compound called delta-aminolevulinic acid (ALA). In subsequent steps, seven other enzymes produce and modify compounds that ultimately lead to heme.
The genes related to porphyria provide instructions for making the enzymes needed to produce heme. Mutations in most of these genes reduce enzyme activity, which limits the amount of heme the body can produce. As a result, compounds called porphyrins and porphyrin precursors, which are formed during the process of heme production, can build up abnormally in the liver and other organs. When these substances accumulate in the skin and interact with sunlight, they cause the cutaneous forms of porphyria. The acute forms of the disease occur when porphyrins and porphyrin precursors build up in and damage the nervous system.
One type of porphyria, porphyria cutanea tarda, results from both genetic and nongenetic factors. About 20 percent of cases are related to mutations in the UROD gene. The remaining cases are not associated with UROD gene mutations and are classified as sporadic. Many factors contribute to the development of porphyria cutanea tarda. These include an increased amount of iron in the liver, alcohol consumption, smoking, hepatitis C or HIV infection, or certain hormones. Mutations in the HFE gene (which cause an iron overload disorder called hemochromatosis) are also associated with porphyria cutanea tarda. Other, as-yet-unidentified genetic factors may also play a role in this form of porphyria.
Frequency of Porphyria
Friday - July 13, 2018 @ 12:50:58
The exact prevalence of porphyria is unknown, but it probably ranges from 1 in 500 to 1 in 50,000 people worldwide. Overall, porphyria cutanea tarda is the most common type of porphyria. For some forms of porphyria, the prevalence is unknown because many people with a genetic mutation associated with the disease never experience signs or symptoms.
Acute intermittent porphyria is the most common form of acute porphyria in most countries. It may occur more frequently in northern European countries, such as Sweden, and in the United Kingdom. Another form of the disorder, hereditary coproporphyria, has been reported mostly in Europe and North America. Variegate porphyria is most common in the Afrikaner population of South Africa; about 3 in 1,000 people in this population have the genetic change that causes this form of the disorder.
Have you tried any of these fantastic tips for Acute Porphyria? AIP, HCP, VP Access to Care Toolkit
Friday - July 13, 2018 @ 07:00:02
I have enjoyed using this ToolKit. Have you tried any of these fantastic tips for Acute Porphyria? AIP, HCP, VP Access to Care Toolkit
Next week we will go through each one to discuss how you can use.
Instructions:
This downloadable Access to Care Toolkit is a resource designed to help patients living with an Acute Porphyria or their caregivers, loved ones and healthcare providers secure access to Panhematin at their preferred health facility. We have recently learned of patients who are being denied this treatment from some hospitals and directed to secure another healthcare provider. If this has happened to you or someone you know or care for, please use these tools to request help from your state and local representatives and health advocacy organizations.
We understand the debilitating effects of Acute Porphyrias and we hope these resources will help you secure access to Panhematin when you need it most.
The toolkit contains the following materials:
Healthcare Conversation Tracker
Customizable letter templates
AIP, HCP, VP Access to Care Fact Sheet
Patient Bill of Rights (Example)
Healthcare Conversation Tracker: Use this simple form to record your experiences and conversations with your doctors, nurses, insurance providers or administrative staff at the hospital. This will give you a clear record of what happened and support your access to care claims.
Customizable letters templates: Download the customizable letter template that pertains to you (as a patient, caregiver, or healthcare provider) and insert your personal details. You can send the letter to various stakeholders including:
Hospital administrator at the hospital that discontinued treatment
Local Ombudsman (a person that investigates and helps settle complaints)
Local and/or State Representatives (e.g., Congressman, Senators)
State Department of Public Health
Insurance Provider
Note: The letter provided is only a sample providing suggestions to the writer for composing his/her own letter. It is the writers responsibility to detail his/her own thoughts and experiences in a personally acceptable manner. All content is ultimately the responsibility of the writer.
AIP, HCP, VP Access to Care Fact Sheet: This fact sheet defines Acute Porphyrias, its symptoms and why its important for patients to get immediate access to approved treatments when necessary. Include this with your letter as an informational supplement for your recipient.
Patient Bill of Rights: Some states have a Patient Bill of Rights that can be used to support your appeal for access to treatment. Included in this toolkit is the Florida Patient Bill of Rights but they can also be found in MN, NJ, NC, and NY. If you dont know whether your state has a Patient Bill of Rights or where to find it, call and ask your state or local public health office.
We hope this toolkit will help you to receive the treatment that you need and deserve.
If you have questions, please contact the American Porphyria Foundation at 1866.APF.3635 or porphyrus@porphyriafoundation.com.
What is Porphyria?
Wednesday - July 11, 2018 @ 07:00:09
Porphyria is a group of disorders caused by abnormalities in the chemical steps that lead to heme production. Heme is a vital molecule for all of the body's organs, although it is most abundant in the blood, bone marrow, and liver. Heme is a component of several iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood).
Researchers have identified several types of porphyria, which are distinguished by their genetic cause and their signs and symptoms. Some types of porphyria, called cutaneous porphyrias, primarily affect the skin. Areas of skin exposed to the sun become fragile and blistered, which can lead to infection, scarring, changes in skin coloring (pigmentation), and increased hair growth. Cutaneous porphyrias include congenital erythropoietic porphyria, erythropoietic protoporphyria, hepatoerythropoietic porphyria, and porphyria cutanea tarda.
Other types of porphyria, called acute porphyrias, primarily affect the nervous system. These disorders are described as "acute" because their signs and symptoms appear quickly and usually last a short time. Episodes of acute porphyria can cause abdominal pain, vomiting, constipation, and diarrhea. During an episode, a person may also experience muscle weakness, seizures, fever, and mental changes such as anxiety and hallucinations. These signs and symptoms can be life-threatening, especially if the muscles that control breathing become paralyzed. Acute porphyrias include acute intermittent porphyria and ALAD deficiency porphyria. Two other forms of porphyria, hereditary coproporphyria and variegate porphyria, can have both acute and cutaneous symptoms.
The porphyrias can also be split into erythropoietic and hepatic types, depending on where damaging compounds called porphyrins and porphyrin precursors first build up in the body. In erythropoietic porphyrias, these compounds originate in the bone marrow. Erythropoietic porphyrias include erythropoietic protoporphyria and congenital erythropoietic porphyria. Health problems associated with erythropoietic porphyrias include a low number of red blood cells (anemia) and enlargement of the spleen (splenomegaly). The other types of porphyrias are considered hepatic porphyrias. In these disorders, porphyrins and porphyrin precursors originate primarily in the liver, leading to abnormal liver function and an increased risk of developing liver cancer.
Environmental factors can strongly influence the occurrence and severity of signs and symptoms of porphyria. Alcohol, smoking, certain drugs, hormones, other illnesses, stress, and dieting or periods without food (fasting) can all trigger the signs and symptoms of some forms of the disorder. Additionally, exposure to sunlight worsens the skin damage in people with cutaneous porphyrias.
Oscar Mannson XLP story of a little man!
Tuesday - July 10, 2018 @ 12:04:48
Oscar Mannson
Oscar was born in London almost 3 years ago, and we stayed in the UK for 2 of the first years of his life. He was a healthy baby and almost never cried. The first time we discovered that he had some similar reactions to the sun as I had was when he was just under 1. We were staying at my partner' fathers summer house that is in Iceland in a wooden cottage. It was a very hot August day, and we had mostly been inside and in the village and only made one short trip to the beach.
In the evening, Oscar was crying and uncomfortable - we had never seen him like this - and we pulled out all the stops. But as he was rubbing his hands and I was feeling kind hot and that tingling feeling in my hands, I thought maybe he did as well. Jonas thought it was strange ( I had not been diagnosed ) but it seemed to help if I did the things that worked for me - so cold water and when I sat with him on the back of the house where it was cold he seemed to be fine. We left the house a day early due to the sleepless nights. We described what had happen to Jonas dad who is a doctor but he could not relate it to anything.
The following year we took a holiday to Menorca, Spain. We left early in the year to avoid strong sun. But when we got there it was hotter than normal. And Oscar started to react like he did in the summer house and again I put cold water and I sat with him on the balcony. We were debating whether to go back - We had a doctor come to the hotel but he just thought Oscar was over tired. We decided to stay we just avoided the sun - we went out early morning late afternoon. But now I was sure that Oscar was reacting the same was as I did - where it feels like youre on fire if you are exposed to the sun. When we got back to the UK I was detriment to do something and I went to our doctors office and I think spoke to 5 Doctors who just took no interest. So in my mind I thought we just had some sun allergies or something. We then moved back to Sweden, and I tried to go to Doctors here and we got referrals to the skin clinic but they said that what I described did not exist.
In May this year, Oscar got a spot at a school. I had told the teachers that he was really sensitive to the sun. it was a really hot May and Oscar got sick right away - Jonas had to go and pick him up they had been out playing in the sun and it was really hot inside as the air con was not working . When they got home Oscar sick and he had burn marks on his hands. It was awful. But Jonas said that one of the teachers had said that she thought that Oscar could have this rare disease that her children had and she would bring the papers to the School. The next day I went to pick up the papers - and it was like I had not been crazy for 30 years there really was a disease and other people with same experience. I started reading everything and was of course sad and worried but also relived as at least now we would know.
To make a long story short it was then really difficult for us to get a referral to get diagnosed - even with papers and Oscars burnt hands the doctors were skeptical and once I got a referral they did not think it was urgent and we should wait until after the summer. I took matters in my own hands and got a blood test from the lab. We have now gotten some help from a very nice professor in Stockholm and we are getting the medical help we need. The results shows that we have x linked EPP - my son has a very high count which is very stressful and we dont know how his life will be as we are the only case in Sweden. There is no one to talk to about it - someone with real experience.
Theres Another Opioid Crisis We Dont Talk About And Im Trapped In The Middle Of It
Saturday - July 7, 2018 @ 07:30:25
Theres Another Opioid Crisis We Dont Talk About And Im Trapped In The Middle Of It
COURTESY OF RICK LUNKENHEIMERRick Lunkenheimer lying at a 45-degree angle on a wedge pillow with extra pillow for neck support, configurable desk and microphone for voice dictation.
I am an opioid user.
There, I said it. Im out of the closet, per se.
I have been fighting chronic back pain for the past 18 years and have been using opioids for the past eight. I am not addicted to the medication and do not take any more than my doctor prescribes.
I am always cognizant of the threat that I may one day become addicted, or worse, that my tolerance levels will rise and I will need more and more pills to receive the same amount of relief. I have to fight off addiction, but more importantly, I need to fight off pain.
The debate on the opioid crisis often overlooks chronic pain patients who take these medications responsibly. Many of us long for an affordable alternative to opioids, which have many adverse side effects.
I have a sitting disability, which means I cannot sit for long periods of time without severe pain, due to lumbago (low back pain) and sciatica (pain that shoots down my left leg). I also have fibromyalgia, which hits different areas of the body at different times. It is difficult for me to get around, sit for dinner, visit family and friends, or sometimes tie my shoes. I am the very definition of a chronic pain patient.
My doctor first prescribed me an opioid in 2010, after back surgery (lumbar fusion at the L5-S1 level). The decision for me to undergo surgery was not taken lightly by my doctor or myself. I spent 10 years attempting to fight the pain in other ways: chiropractors, physical therapists, acupuncture, acupressure, dry needling, yoga, meditation, massage, biofeedback, and steroid and collagen injections. The surgery was a last resort. So were the opioids. I had tried every other non-opioid pain medication on the market. The back surgery was a failure, and my doctor decided to keep me on them.
Opioids are not my only tool to fight pain. My preferred method is to lie on large ice packs and let the weight of my body sink into them. I also use heating pads, a transcutaneous electrical nerve stimulation unit, sprays, patches, and just avoiding sitting or standing for too long because it aggravates pain. Sprays and patches are smelly, TENS units are cumbersome, heating pads need electrical sockets, and ice packs require me to lie down, which is hard in public. Opioids are not a magic bullet, but they are portable when other forms are not reasonable.
At home, I can control my pain with fewer pain medications, but this leads to a cycle of isolation that many chronic pain patients experience. I am afraid to travel because it is difficult to sit in public places, so I tend to stay at home, sometimes days at a time. My doctors want me to live my life, even if it means taking the full dosage of pain medication. They are more worried about my isolation than my threat of addiction.
COURTESY OF RICK LUNKENHEIMERLunkenheimers workstation outfitted to help manage his pain with a sit-to-stand desk with raised monitor for correct position, left-hand trackpad for scrolling up and down, right-hand mouse for clicking, and anti-fatigue pad for standing. If sitting, he uses an ergonomic chair with wedge pillow to raise his hips above his knees and an inflatable pillow for lumbar support.
Patients like me live in fear of further crackdowns on opioid prescriptions. In October 2014, the Drug Enforcement Administration changed the medication I used, hydrocodone, to a Schedule II drug. At the time, I had been seeing my doctor once every two months for a checkup and a prescription that included another refill. After the change, hydrocodone could not legally be refilled without a brand-new paper script, so patients now needed an appointment each month.
I doubled the number of appointments with my pain specialist per year. It may seem like a small inconvenience for the patient, but this required pain specialists to schedule double or triple the number of appointments each month to take care of prescriptions for all of their patients.
Subscribe to Must Reads
The internet's best stories, and interviews with their authors.
Thus, slots with the doctor became a scarce resource, and the net result was that many chronic pain patients werent able to get their medications as all pain doctors were swamped during the transition. Other doctors were not willing to give opioids to a patient who was new to them, even if the patient had been on opioids from another doctor for years.
Many chronic pain patients have already gone the extra mile to fight the illnesses that inflict us with both Western and Eastern treatments. We have tried things that have not helped and have not tried things that are too expensive and not covered by health insurance. We do not mind peeing in a cup to help fight the opioid crisis that has afflicted others. Instead, we worry that new regulations will make more patients ineligible and we will not make the cut, despite using these medications for years. We fear repeal-and-dont-replace fixes, where the government restricts opioids but alternatives are still not covered by insurance.
Opioid use is often lumped together with heroin use because opioids are synthetic versions of the poppy seed used to create heroin and can have similar effects. That is where the similarities end. One is prescribed by doctors for medical purposes, while the other is a street drug used recreationally. Heroin destroys lives. Opioids can also ruin lives if prescribed and used recklessly, but for most people, it can give them some of their life back. For instance, it allows me to get out of the house once in a while.
In the war on illegal drugs, it is essential to fight from both ends: 1. stop the drug dealers and 2. help those addicted.
Fighting the opioid crisis is similar: 1. place limits on opioid prescriptions (particularly for acute pain) and 2. help those addicted to opioids.
I applaud these efforts. However, because opioids are not designed to be recreational drugs, there is a third aspect to this war concerning chronic pain. We need to fight the medical cause as well. In short, we must be willing to pay for more advanced (and expensive) medical treatments.
It is not acceptable to stop treating chronic pain. It is not acceptable to tell people to just live with it. It is real and affects peoples lives in catastrophic ways.
Anti-opioid advocates must be pro-something else. Often, they point to alternative medicines, like acupuncture, yet do not call for health insurance companies pay for these more expensive treatments. They talk about the benefits of medical cannabis in replacing opioids for pain management in some patients but dont call for nationwide legalization, let alone requiring insurance companies to pay for it. Advanced pain management centers have had success in getting patients off opioids, but they are well beyond the financial reach of most people.
It is not acceptable to stop treating chronic pain. It is not acceptable to tell people to just live with it. It is real and affects peoples lives in catastrophic ways. Many who have experienced acute pain, such as from an accident or surgery, still dont understand how chronic illness affects an individual. Chronic pain is not merely acute pain for an extended period of time. Long-term pain changes the patients lifestyle and isolates them. The knowledge that chronic pain will never get better prevents patients from living their lives to the fullest.
Opioids and other painkillers are not meant to cure diseases or fix structural problems. They are there to manage pain. For myself, I am not expecting a cure. My only goal is to control the pain from day to day. We cannot deny that our country is facing a dangerous opioid crisis, but we also have to recognize the positive effect opioids have on those that need them the most. We cannot ignore the pain that leads to opioid prescriptions in the first place. We must be willing to accept that the cost of solving the opioid crisis is a higher cost of health care. Is that a pill we are willing to swallow?
When friends and family talk to me about my sitting disability, I often hear sentences that start with, Have you tried and the sentence ends with something I had either tried and didnt work, or would like to try but it is not covered by my insurance. When looking for new treatments to manage my pain, I no longer think in terms of tradition versus alternative medicine, or Eastern versus Western approaches. My concern is: Will my insurance company pay for this, or wont it? I read all sorts of stories about groundbreaking treatments, but none of them are covered by insurance or within my grasp.
I would love to stop taking opioids. Trust me; I would. The side effects are almost as severe as the pain. But I have not found a suitable alternative. My sitting disability makes sitting painful. Therefore, I tend to stay home when I can, avoiding social interactions and daily activity most people take for granted.
Taking my pain medication can help me to sit through a dinner or a movie. It can help me spend six hours out of the house, rather than only four. The medication wont remove the pain altogether, but it might reduce it from a pain level 7 to a pain level 3. Its the best I can do without adequate pain management.
Opioids can be dangerous for some, but for me, they have helped me merely to live my life.
The Opioid problem is real! Please have your questions written down before you speak to a physician about your pain. Even draw and journal how long you have had it.
Please be balanced in all things. Good health to you!
Porphyria Post
Monday - July 2, 2018 @ 07:00:08
Request Priority Review of Scenesse
URGENT NEED FOR LETTERS
The new drug application for Scenesse has been submitted to the FDA - and it is time to advocate! We need your letters to the FDA Commisioner requesting Priority Review. Please send your letter immediately to add to our growing pile!! It is starting to look impressive!
All letters must be signed and sent to the APF ASAP.
We will submit the letters to the FDA by July 19, 2018.
Begin your letter:
Scott Gottlieb, M.D.
FDA Commissioner
U.S. Food and Drug Administration
10903 New Hampshire Avenue
Silver Spring, MD 20993
Dear FDA Commissioner:
Mail your letter to:
American Porphyria Foundation
4915 St. Elmo Avenue, Suite 105
Bethesda, Maryland 20814
Alternatively you can scan your signed letter and email it to porphyrus@porphyriafoundation.com.
We will present all of the letters at once to the FDA!
Please send letters today or ASAP! Time is of the Essence!
Clinical Trial for EPP!
Research is the key to your cure! What is the most important element in research? YOU! You now have an opportunity to participate in a trial for a new EPP investigational drug. Contact the APF for details. YOU are our hope for the future. Contact the APF on 1-866-APF-3635 or email porphyrus@porphyriafoundation.com.
An oral investigational drug has been developed with the potential to increase sunlight duration and tolerance in individuals with EPP. This phase II clinical trial is needed to understand if the investigational drug works and to ensure that it is safe. The study duration is 24 weeks including follow-up. You must be age 18-70 to participate
and have a confirmed diagnosis of EPP. All travel is included and will be arranged by a concierge service.
"RememberResearch is the key to YOUR cure!
Each Step Toward Finding an Effective Treatment is Important!
Givosiran Trials
Thanks to YOU, our APF members and patients, the Givosiran trials are now full. We are so proud of each of you for participating in this important research. As of June 29, 2018 the trials have closed for enrollment.
Update your records.
We have moved.
The American Porphyria Foundation is excited to announce out move to Bethesda, Maryland - next door to our nation's capital. Please update your records with our new address.
American Porphyria Foundatin
4915 St. Elmo Ave., Suite 105
Bethesda, MD 20814
Telephone: 301.347.7166
porphyrus@porphyriafoundation.com
Rare Disease Legislative Advocates is hosting In-District Lobby Days to facilitate meetings for rare disease advocates across the country with members of Congress in their local offices during the 2018 summer in-district work period (July 31st through Sept. 4th).
Meeting with your representative and senators throughout the year is critical to building key relationships. These meetings also provide an opportunity to discuss legislation that is meaningful to the rare disease community, and to highlight the importance of the Rare Disease Congressional Caucus. Make YOUR voice heard as you advocate on behalf of the rare disease community!
Registration for In-District Lobby Days is FREE and open through July 4th. You will be provided the option to specify you availability and the distance you are willing to travel for a meeting.
All In-District Lobby Day participants will be invited to a webinar on Thursday, July 26th,at 2pm EST to go over key legislative issues and tips for successful meetings. Legislative asks will be available RDLA's website by late July.
Contact Information
Is your contact Information up to date?
If not please give us a call @ 866-APF-3635 or Email to porphyrus@porphyriafoundation.com
American Porphyria Foundation 1.866.APF.3635 porphyriafoundation.org
STAY CONNECTED
"Remember.Research is the Key to Your Cure!"
The Patient - Patient-Centered Outcomes Research https://doi.org/10.1007/s40271-018-0319-3
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
References 1. Puy H, Gouya L, Deybach JC. Porphyrias. Lancet. 2010;375(9718):92437. 2. Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142(6):43950. 3. Balwani M, Desnick RJ. The porphyrias: advances in diagnosis and treatment. Blood. 2012;120(23):4496504. 4. Pischik E, Kauppinen R. An update of clinical management of acute intermittent porphyria. Appl Clin Genet. 2015;8:20114. 5. Bonkovsky HL, Maddukuri VC, Yazici C, Anderson KE, Bissell DM, Bloomer JR, et al. Acute porphyrias in the USA: features of 108 subjects from porphyrias consortium. Am J Med. 2014;127(12):123341. 6. Stein P, Badminton M, Barth J, Rees D, Stewart MF, et al. Best practice guidelines on clinical management of acute attacks of porphyria and their complications. Ann Clin Biochem. 2013;50(Pt 3):21723. 7. Jeans JB, Savik K, Gross CR, Weimer MK, Bossenmaier IC, Pierach CA, et al. Mortality in patients with acute intermittent porphyria requiring hospitalization: a United States case series. Am J Med Genet. 1996;65(4):26973. 8. Elder G, Harper P, Badminton M, Sandberg S, Deybach JC. The incidence of inherited porphyrias in Europe. J Inherit Metab Dis. 2013;36(5):84957. 9. Naik H, Stoecker M, Sanderson SC, Balwani M, Desnick RJ. Experiences and concerns of patients with recurrent attacks of acute hepatic porphyria: a qualitative study. Mol Genet Metab. 2016;119(3):27883. 10. Millward LM, Kelly P, Deacon A, Senior V, Peters TJ. Self-rated psychosocial consequences and quality of life in the acute porphyrias. J Inherit Metab Dis. 2001;24(7):73347. 11. Andersson C, Innala E, Backstrom T. Acute intermittent porphyria in women: clinical expression, use and experience of exogenous sex hormones: a population-based study in northern Sweden. J Intern Med. 2003;254(2):17683. Patient Perspective on Acute Intermittent Porphyria with Frequent Attacks 12. Gouya L. EXPLORE: a prospective, multinational natural history study of acute hepatic porphyria patients with recurrent attacks. International Liver Congress, Paris, 1115 Apr 2018. 13. Marsden JT, Guppy S, Stein P, Cox TM, Badminton M, Gardiner T, et al. Audit of the use of regular haem arginate infusions in patients with acute porphyria to prevent recurrent symptoms. JIMD Rep. 2015;22:5765. 14. Millward LM, Kelly P, King A, Peters TJ. Anxiety and depression in the acute porphyrias. J Inherit Metab Dis. 2005;28(6):1099107. 15. Jimenez-Monreal AM, Murcia MA, Gomez-Murcia V, Bibiloni Mdel M, Pons A, Tur JA, et al. Anthropometric and quality-oflife parameters in acute intermittent porphyria patients. Medicine (Baltimore). 2015;94(30):e1023. 16. Wikberg A, Jansson L, Lithner F. Womens experience of suffering repeated severe attacks of acute intermittent porphyria. J Adv Nurs. 2000;32(6):134855. 17. Britten N. Qualitative interviews in medical research. BMJ. 1995;311(6999):2513. 18. US Food and Drug Administration. Patient-reported outcome measures: use in medical product development to support labelling claims. 2009. https ://www.fda.gov/downl oads/drugs /guida nces/ucm19 3282.pdf. Accessed 7 Jun 2018. 19. US Food and Drug Administration. Roadmap to patient-focused outcome measurement in clinical trials. 2013. https ://www.fda. gov/Drugs /Devel opmen tAppr ovalP roces s/DrugD evelo pment Tools Quali ficat ionPr ogram /ucm37 0177.htm. Accessed 7 Jun 2018. 20. Thomas DR. A general inductive approach for analyzing qualitative evaluation data. Am J Eval. 2006;27(2):23746. 21. Bryman A, Burgess B. Analyzing qualitative data. New York: Routledge; 2002. 22. Mason M. Sample size and saturation in PhD studies using qualitative interviews. Forum Qual Soc Sci Res. 2010. https ://doi. org/10.17169 /fqs-11.3.1428 23. Brod M, Tesler LE, Christensen TL. Qualitative research and content validity: developing best practices based on science and experience. Qual Life Res. 2009;18(9):126378. 24. American Porphyria Foundation. The voice of the patient: patient-focused drug development meeting. Acute porphyrias. 2017. http://www.porph yriaf ounda tion.com/sites /defau lt/files /APFPF DDMee tingM arch1 %2C201 7-TheVo iceof thePa tient %281%29.pdf. Accessed 7 Jun 2018. 25. Bylesjo I, Wikberg A, Andersson C. Clinical aspects of acute intermittent porphyria in northern Sweden: a population-based study. Scand J Clin Lab Invest. 2009;69(5):6128. 26. Hyland ME, Lanario JW, Pooler J, Masoli M, Jones RC. How patient participation was used to develop a questionnaire that is fit for purpose for assessing quality of life in severe asthma. Health Qual Life Outcomes. 2018;16:24. 27. Carlsen B, Glenton C. What about N? A methodological study of sample-size reporting in focus group studies. BMC Med Res Methodol. 2011;11:26. https ://doi.org/10.1186/1471-2288-11-26.
Family, Friends, Spouse of an EPP Hero Please Write FDA a Letter
Friday - June 29, 2018 @ 10:20:36
THANK YOU TO ALL FOR YOUR LETTERS REQUESTING PRIORITY REVIEW FOR SCENESSE! They are starting to pour into the APF office. Haven't taken a moment to write yetnow is the time. Don't delay.
ADDRESS TO: Scott Gottlieb, M.D. FDA Commissioner U.S. Food and Drug Administration 10903 New Hampshire Avenue Silver Spring, MD 20993 Dear FDA Commissioner:
MAIL TO: American Porphyria Foundation 4915 St. Elmo Avenue, Suite 105 Bethesda, Maryland 20814
OR EMAIL TO: porphyrus@porphyriafoundation.com
Porphyria POST regarding Letters & Trial Info
Friday - June 29, 2018 @ 07:00:12
You can be a medical hero! A new investigational drug for EPP is ready for clinical trial. All we need to begin is YOU! The trial spots will fill up fastâ?¦join now! Contact the APF on 1-866-APF-2635 or email porphyrus@porphyriafoundation.orgto be connected to a study site. Dont wait! Rememberâ?¦Research is the Key to Your Cure! An oral investigational drug has been developed with the potential to increase sunlight duration and tolerance in individuals with EPP. This phase II clinical trial is needed to understand if the investigational drug works and to ensure that it is safe. The study duration is 24 weeks including follow-up. You must be age 18-70 to participate and have a confirmed diagnosis of EPP. All travel is included and will be arranged by a concierge service. "RememberResearch is the key to YOUR cure!" Each Step Toward Finding an Effective Treatment is Important!
Porphyria Post
Request
The New Drug Application for Scenesse has been submitted to the FDA.
Now is the time to work hard as patient advocates. The APF is launching a new campaign to request Priority Review
Now is the time to send your letter requesting priority review for Scenesse. We need to move Scenesse, a long-awaited treatment for EPP, through the regulatory process as quickly as possible. Now that a complete new drug application has been submitted to the FDA - they have the option of granting priority review which moves the process faster. Please share with the FDA
* That there is no treatment for EPP * That there is an unmet medical need for a treatment for EPP * That living with EPP is a tremendous burden * How living with EPP affects your quality of life
It is important that you send your signed letter to the APF immediately.
Begin your letter: Scott Gottlieb, M.D. FDA Commissioner U.S. Food and Drug Administration 10903 New Hampshire Avenue Silver Spring, MD 20993 Dear FDA Commissioner:
Mail your letter to: American Porphyria Foundation 4915 St. Elmo Avenue, Suite 105 Bethesda, Maryland 20814
Alternatively you can scan your signed letter and email it to porphyrus@porphyriafoundation.com. All letters must be signed.
We will present all of the letters at once to the FDA! Please send letters today or ASAP! Time is of the Essence!
Study # 7210 Newer Direct-Acting Anti-Viral Agents as Sole Therapy of Porphyria Cutanea Tarda in Subjects with Chronic Hepatitis C
Newer Direct-Acting Anti-Viral Agents as Sole Therapy of Porphyria Cutanea Tarda in Subjects with Chronic Hepatitis C
How to participate:
In order to participate in a study, you must personally contact
the APF Edrin 1/866/APF/3635
For Diseases:
Porphyria Cutanea Tarda (PCT)
Background
Many people with PCT also have hepatitis C virus (HCV). There have been reports that the treatment of HCV infection can also resolve the symptoms of PCT. However, this has not been clearly established.
Therefore, the purpose of this study is to determine if giving a standard treatment for HCV is also an effective treatment for PCT.
The research aims are:
To assess whether Harvoni (the treatment for HCV) alone is an effective and durable treatment for PCT in patients with both HCV and PCT.
To determine whether treating patients with PCT and HCV with Harvoni is as effective as treating PCT with the standard therapies, phlebotomy or hydroxychloroquine.
About this Study
This is a clinical trial, which means its purpose is to study an intervention or treatment. In this study all patients with PCT will be given a standard dose of Harvoni and monitored for two years. Currently there are two standard therapies for PCT, phlebotomies (removing certain amounts of blood at specific intervals), or low dose hydroxychloroquine (an oral pill). These treatments are used for patients with PCT whether or not they also have HCV. For patients with HCV however, we do not know whether treating the HCV first will also resolve the PCT symptoms.
There will be an initial visit to determine whether participants are eligible to be in the study. If a participant is found to be eligible, he/she will be asked come to the study site once every month over the course of one year, and then once every 3 months for an additional year. There will be approximately 17 visits over the course of the whole study. At these visits the study doctors will check in with the participant and some blood and urine samples will be taken. Participants will not be charged for any of the lab tests that are being done as a part of this study alone. All participants in this study will receive the Harvoni pills at no cost to them.
Targeted Enrollment
To be eligible to participate, you must:
Have a biochemical diagnosis of Porphyria Cutanea Tarda (PCT)
Have the typical symptoms of PCT (blistering in response to sun exposure, fragile skin, etc.)
Be at least 18 years old
Have chronic hepatitis C virus (HCV)
If you are female of child-bearing potential, you must be willing to take an effective method of contraception
You are not eligible to participate if you:
Are pregnant, planning to become pregnant, or breast-feeding
Used any other clinical or experimental therapy for PCT in the past month
Are currently enrolled in the Porphyrias Consortium study Therapeutic Studies of PCT
Have an HIV infection that is not well controlled
Drink more than 25 alcoholic drinks in a week
Have been taking a medication called amiodarone within 60 days of enrolling in this study
Have been taking St. Johns wort within 28 days or enrolling in this study
Have uncontrolled diabetes
Have chronic hepatitis B
Have other liver disease or have liver cancer
Have had a liver transplant in the past
Another disease or condition that the study doctors judge would interfere with the study
How to participate:
In order to participate in a study, you must personally contact the
PORPHYRIA POST URGENT
Wednesday - June 27, 2018 @ 10:40:32
Porphyria Post
Request
The New Drug Application for Scenesse has been submitted to the FDA.
Now is the time to work hard as patient advocates. The APF is launching a new campaign to request Priority Review
Now is the time to send your letter requesting priority review for Scenesse. We need to move Scenesse, a long-awaited treatment for EPP, through the regulatory process as quickly as possible. Now that a complete new drug application has been submitted to the FDA - they have the option of granting priority review which moves the process faster. Please share with the FDA
* That there is no treatment for EPP * That there is an unmet medical need for a treatment for EPP * That living with EPP is a tremendous burden * How living with EPP affects your quality of life
It is important that you send your signed letter to the APF immediately.
Begin your letter: Scott Gottlieb, M.D. FDA Commissioner U.S. Food and Drug Administration 10903 New Hampshire Avenue Silver Spring, MD 20993 Dear FDA Commissioner:
Mail your letter to: American Porphyria Foundation 4915 St. Elmo Avenue, Suite 105 Bethesda, Maryland 20814
Alternatively you can scan your signed letter and email it to porphyrus@porphyriafoundation.com. All letters must be signed.
We will present all of the letters at once to the FDA! Please send letters today or ASAP! Time is of the Essence!
Research is the key to your cure!
Wednesday - June 27, 2018 @ 07:00:05
Research is the key to your cure! What is the most important element in research? YOU! You now have an opportunity to participate in a trial for a new EPP investigational drug. Contact the APF for details. YOU are our hope for the future. Contact the APF on 1-866-APF-3635 or email porphyus@porphyriafoundation.org. An oral investigational drug has been developed with the potential to increase sunlight duration and tolerance in individuals with EPP. This phase II clinical trial is needed to understand if the investigational drug works and to ensure that it is safe. The study duration is 24 weeks including follow-up. You must be age 18-70 to participate and have a confirmed diagnosis of EPP. All travel is included and will be arranged by a concierge service. "RememberResearch is the key to YOUR cure! Each Step Toward Finding an Effective Treatment is Important!
7209 GET INVOLVED STUDIES 7209 Effect of Oral Iron Therapy on Erythrocyte Protoporphyrin Levels in the Erythropoietic Protoporphyrias
Effect of Oral Iron Therapy on Erythrocyte Protoporphyrin Levels in the Erythropoietic Protoporphyrias
How to participate:
In order to participate in a study, you must personally contact
the APF Edrin 1/866/APF/3635
For Diseases:
Erythropoietic Protoporphyria (EPP)
X-Linked Protoporphyria (XLP)
Background
In the medical literature, there are conflicting reports on whether iron improves symptoms in patients with EPP or XLP. Giving iron to people who are iron deficient is thought to improve EPP symptoms; however, this has never been systematically tested.
Therefore, the purpose of this study is to determine the effect of oral iron on EPP and XLP patients.
The research aims are:
To determine if oral iron can decrease erythrocyte protoporphyrin levels in patients with EPP or XLP
To assess whether oral iron can improve iron status and decrease plasma porphyrin levels and symptoms in patients with EPP or XLP
To assess if there is an improvement in the quality of life in treated patients
About this Study
This is a clinical trial, which means its purpose is to study an intervention or treatment. In this study all patients with EPP or XLP will be given a standard dose of iron pills and monitored for one year. There is currently no effective Food and Drug Administration (FDA) approved treatment for EPP or XLP in the US. Giving iron to patients with low ferritin (a measure of body iron stores) levels may help improve their EPP symptoms by decreasing erythrocyte protoporphyrin levels.
Participants will be asked come to the study site once every three months over the course of one year for a total of five visits. At these visits the study doctors will check in with the participant and some blood and urine samples will be taken. Participants will not be charged for any of the lab tests that are being done as a part of this study alone. In between each study visit, there will be one phone call to check in and see how each participant is doing. All patients in this study will receive iron pills at no cost to them.
Have a biochemical diagnosis of erythropoietic protoporphyria (EPP) or X-Linked Protoporphyria (XLP)
Be at least 18 years old
Have a serum ferritin level of less than or equal to 30 ng/mL
You are not eligible to participate if you have:
Had a liver or bone marrow transplant, or have significant liver disease determined by the study doctor
A known or suspected allergy to oral iron
Used any other clinical or experimental therapy in the past 3 months
Another disease or condition that the study doctors judge would interfere with the study
How to participate:
In order to participate in a study, you must personally contact the
EPP Clinical Trial
Monday - June 25, 2018 @ 12:05:51
You can be a medical hero! A new investigational drug for EPP is ready for clinical trial. All we need to begin is YOU! The trial spots will fill up fastâ?¦join now! Contact the APF on 1-866-APF-2635 or email porphyrus@porphyriafoundation.orgto be connected to a study site. Dont wait! Rememberâ?¦Research is the Key to Your Cure!
An oral investigational drug has been developed with the potential to increase sunlight duration and tolerance in individuals with EPP. This phase II clinical trial is needed to understand if the investigational drug works and to ensure that it is safe. The study duration is 24 weeks including follow-up. You must be age 18-70 to participate and have a confirmed diagnosis of EPP. All travel is included and will be arranged by a concierge service. "RememberResearch is the key to YOUR cure!" Each Step Toward Finding an Effective Treatment is Important!
Porphyria Post IMPORTANT
Monday - June 25, 2018 @ 07:00:06
Porphyria Post
Request
The New Drug Application for Scenesse has been submitted to the FDA.
Now is the time to work hard as patient advocates. The APF is launching a new campaign to request Priority Review
Now is the time to send your letter requesting priority review for Scenesse. We need to move Scenesse, a long-awaited treatment for EPP, through the regulatory process as quickly as possible. Now that a complete new drug application has been submitted to the FDA - they have the option of granting priority review which moves the process faster. Please share with the FDA
* That there is no treatment for EPP * That there is an unmet medical need for a treatment for EPP * That living with EPP is a tremendous burden * How living with EPP affects your quality of life
It is important that you send your signed letter to the APF immediately.
Begin your letter: Scott Gottlieb, M.D. FDA Commissioner U.S. Food and Drug Administration 10903 New Hampshire Avenue Silver Spring, MD 20993 Dear FDA Commissioner:
Mail your letter to: American Porphyria Foundation 4915 St. Elmo Avenue, Suite 105 Bethesda, Maryland 20814
Alternatively you can scan your signed letter and email it to porphyrus@porphyriafoundation.com. All letters must be signed.
We will present all of the letters at once to the FDA! Please send letters today or ASAP! Time is of the Essence!
Update your records.
We have moved.
The American Porphyria Foundation is excited to announce out move to Bethesda, Maryland - next door to our nation's capital. Please update your records with our new address.
American Porphyria Foundatin 4915 St. Elmo Ave., Suite 105 Bethesda, MD 20814 Telephone: 301.347.7166 porphyrus@porphyria.com
Rare Disease Legislative Advocates is hosting In-District Lobby Days to facilitate meetings for rare disease advocates across the country with members of Congress in their local offices during the 2018 summer in-district work period (July 31st through Sept. 4th).
Meeting with your representative and senators throughout the year is critical to building key relationships. These meetings also provide an opportunity to discuss legislation that is meaningful to the rare disease community, and to highlight the importance of the Rare Disease Congressional Caucus. Make YOUR voice heard as you advocate on behalf of the rare disease community!
Registration for In-District Lobby Days is FREE and open through July 4th. You will be provided the option to specify you availability and the distance you are willing to travel for a meeting.
All In-District Lobby Day participants will be invited to a webinar on Thursday, July 26th,at 2pm EST to go over key legislative issues and tips for successful meetings. Legislative asks will be available RDLA's website by late July.
Get Involved
â?¢ Research â?¢
â?¦Donateâ?¦
â?¢ Visit our Website â?¢ Contact Information
Is your contact Information up to date? If not please give us a call @ 866-APF-3635 or Email to porphyrus@porphyriafoundation.com
Watch out, Boise Spartan Race, here comes Jared Ulmer! A self-described family man and health enthusiast, Jared also lives with Erythropoietic Protoporphyria (EPP) - a rare diseases that results in extreme pain with exposure to sunlight. You may know Jared from his YouTube channel, PorphyriaJ where he creates awareness about this rare disease and illustrates living with it in his own unique way through honest and entertaining short videos. (Porphyria J)
This Saturday JUNE 23rd at 8:45AM MST, Jared will take on the SPARTAN SPRINT! Jared will run 3+ miles and overcome 23 daunting obstacles in the Boise Spartan Sprint on behalf of Porphyrians everywhere. The Spartan race motivates people to get out of their comfort zones and grow through resiliency. In order for Jared to complete this race - he will test every ounce of his resilience. For Jared, this race is an outward expression of a deep internal struggle that has developed through a lifelong challenge of avoiding sun exposure. He wants all who suffer from this disease - which has no FDA-approved treatment and no cure - to know they are not alone and to believe that positivity is created by taking care of yourself and focusing on what you can do. You will hear Jared say 'Seek Shade and Stay Happy' - a motto he lives by and shares with all who suffer along with him.
Jared will race on behalf of the AMERICAN PORPHYRIA FOUNDATION. Donations to the American Porphyria Foundation can be made through FirstGiving. Please help Jared reach his goal of raising $1,000. The APF enhances public awareness, develops education programs and materials, and supports research to improve treatment and ultimately a cure for Porphyria. Jared still has his original APF pamphlet and free materials he and his parents first received due to donations like yours. These free materials are what educated Jared on how to handle EPP, and that let him know he wasn't alone. PLEASE SUPPORT JARED AND ALL WHO LIVE WITH EPP ON JUNE 23RD BY DONATING TO THE APF!
GET INVOLVED STUDIES 7207 Erythropoietic Protoporphyrias: Studies of the Natural History, Genotype-Phenotype Correlations, and Psychosocial Impact)
Erythropoietic Protoporphyrias: Studies of the Natural History, Genotype-Phenotype Correlations, and Psychosocial Impact)
How to participate:
In order to participate in a study, you must personally contact the
APF Edrin 1-866-APF-3635
Status: Recruiting
Background
The Erythropoietic Protoporphyrias include EPP and XLP. (Together they are the third most common porphyria and the most common in children.
The purpose of this study is to learn more about EPP and XLP and how to better treat people with them.
Researchers will observe participants over several years to learn about:
How often certain signs that indicate how bad the disease is occur as they are related to skin symptoms. For example, levels of protoporphyrins effects on the liver and other tissues affected by erythropoietic protoporphyrias.
What are the psychosocial effects of living with EPP/XLP.
About this Study
This is a longitudinal study (study taking place over 5 years) of about 150 individuals with EPP. Participants enrolled in this study should also be enrolled in the Longitudinal Study of the Porphyrias. Those participating in this study will be evaluated yearly. If possible, the first study visit should happen at one of the Porphyrias Consortium clinical sites (see the list of participating sites below).
Participation in this study will involve the following:
Accessing the information that you provided for the Longitudinal Study of the Porphyrias, including the results of the physical examinations, medical history, family history, questionnaires, and the results of laboratory tests;
Review of your medical records at each study visit; the study doctor may request medical records in between study visits to see how you are doing or if there is a medical concern that may be related to your porphyria or to the study;
Collection of samples, including:
blood (about 2-4 tablespoons)
urine (about ¼ cup) for testing and storage.
Completion of quality of life questionnaires
Testing on these samples may include measurement of certain products found in the blood and urine of people with porphyria and DNA analysis to find the mutation (or change) in the porphyria gene that is causing your porphyria. These samples will be collected at your first study visit, however, we may ask for additional samples later.
Have a diagnosis of erythropoietic protoporphyria (EPP) or X-Linked Protoporphyria (XLP)
You are not eligible to participate if you have:
You have elevations of porphyrins in urine, plasma or erythrocytes due to other diseases (i.e. secondary porphyrinuria or porphyrinemia), such as liver and bone marrow diseases.
A prior diagnosis of porphyria that cannot be documented by review of existing medical records or repeat biochemical or DNA testing.
Porphyria Post
Thursday - June 21, 2018 @ 13:26:08
Porphyria Post
New EPP Clinical Trials
Clinical Trial for EPP!
Participants are needed for participation in a Phase II clinical trial to study an investigational drug for EPP. CONTACT THE APF ON 1-866-APF-3635 or email porphyrus@porphyria.com to learn more.
An oral investigational drug has been developed with the potential to increase sunlight duration and tolerance in
individuals with EPP. This phase II clinical trial is needed to understand if the investigational drug works and to
ensure that it is safe. The study duration is 24 weeks including follow-up. You must be age 18-70 to participate and have a confirmed diagnosis of EPP. All travel is included and will be arranged by a concierge service.
Contact the APF today trial spaces will fill quickly! 1-866-APF-3635.
"RememberResearch is the key to YOUR cure!"
Each Step Toward Finding an Effective Treatment is Important!
SUPPORT OUR VERY OWN SPARTAN!
Watch out, Boise Spartan Race, here comes Jared Ulmer! A self-described family man and health enthusiast, Jared also lives with Erythropoietic Protoporphyria (EPP) - a rare diseases that results in extreme pain with exposure to sunlight. You may know Jared from his YouTube channel, PorphyriaJ where he creates awareness about this rare disease and illustrates living with it in his own unique way through honest and entertaining short videos. (Porphyria J)
This Saturday JUNE 23rd at 8:45AM MST, Jared will take on the SPARTAN SPRINT! Jared will run 3+ miles and overcome 23 daunting obstacles in the Boise Spartan Sprint on behalf of Porphyrians everywhere. The Spartan race motivates people to get out of their comfort zones and grow through resiliency. In order for Jared to complete this race - he will test every ounce of his resilience. For Jared, this race is an outward expression of a deep internal struggle that has developed through a lifelong challenge of avoiding sun exposure. He wants all who suffer from this disease - which has no FDA-approved treatment and no cure - to know they are not alone and to believe that positivity is created by taking care of yourself and focusing on what you can do. You will hear Jared say 'Seek Shade and Stay Happy' - a motto he lives by and shares with all who suffer along with him.
Jared will race on behalf of the AMERICAN PORPHYRIA FOUNDATION. Donations to the American Porphyria Foundation can be made through FirstGiving. Please help Jared reach his goal of raising $1,000. The APF enhances public awareness, develops education programs and materials, and supports research to improve treatment and ultimately a cure for Porphyria. Jared still has his original APF pamphlet and free materials he and his parents first received due to donations like yours. These free materials are what educated Jared on how to handle EPP, and that let him know he wasn't alone. PLEASE SUPPORT JARED AND ALL WHO LIVE WITH EPP ON JUNE 23RD BY DONATING TO THE APF!
Patient Perspective on Acute Intermittent Porphyria with Frequent Attacks:
A Disease with Intermittent and Chronic Manifestations
A new article was published recently about the patient perspective on Acute Intermittent Porphyria with frequent attacks. Click below to view the entire publication.
The Patient - Patient-Centered Outcomes Research https://doi.org/10.1007/s40271-018-0319-3
Givosiran Trials Deadline
The life changing Givosiran research trials will be closing for more acute porphyria patients on Friday, June 29.
YOU have the opportunity to become a patient volunteer. As such, you will receive FREE DRUG for TWO YEARS. Your travel expenses will be paid, including flights, train, car, meals, hotel and a travel stipend.
We cannot improve treatment or have a cure unless YOU , the patient, volunteer FOR THE RESEARCH. If you have an acute porphyria, please be a medical hero and join the trials in the cities below. You will be with a porphyria expert and a research team. Please call Edrin at the APF Office on 301.347.7166. or our toll free number 866 APF 3635.
Rare Disease Legislative Advocates is hosting In-District Lobby Days to facilitate meetings for rare disease advocates across the country with members of Congress in their local offices during the 2018 summer in-district work period (July 31st through Sept. 4th).
Meeting with your representative and senators throughout the year is critical to building key relationships. These meetings also provide an opportunity to discuss legislation that is meaningful to the rare disease community, and to highlight the importance of the Rare Disease Congressional Caucus. Make YOUR voice heard as you advocate on behalf of the rare disease community!
Registration for In-District Lobby Days is FREE and open through July 4th. You will be provided the option to specify you availability and the distance you are willing to travel for a meeting.
All In-District Lobby Day participants will be invited to a webinar on Thursday, July 26th,at 2pm EST to go over key legislative issues and tips for successful meetings. Legislative asks will be available RDLA's website by late July.
Update your records.
We have moved.
The American Porphyria Foundation is excited to announce out move to Bethesda, Maryland - next door to our nation's capital. Please update your records with our new address.
An oral investigational drug has been developed with the potential to increase sunlight duration and tolerance in individuals with EPP. This phase II clinical trial is needed to understand if the investigational drug works and to ensure that it is safe. The study duration is 24 weeks including follow-up.
You must be age 18-70 to participate and have a confirmed diagnosis of EPP. All travel is included and will be arranged by a concierge service.
Contact the APF today trial spaces will fill quickly! 1-866-APF-3635.
"RememberResearch is the key to YOUR cure!"
Each Step Toward Finding an Effective Treatment is Important!
7206 GET INVOLVED STUDIES 7206 Therapeutic Studies in Porphyria Cutanea Tarda
In order to participate in a study, you must personally
contact EDRIN @ APF 1/866/APF/3635
Status: Recruiting
Background
Porphyria Cutanea Tarda (PCT) is the most common porphyria that is generally diagnosed in adulthood. It is also the most responsive type of porphyria to treat. There are two current treatments for PCT. The first and more common treatment is repeated phlebotomies (removing blood, but that also includes the potential disadvantages of discomfort, inconvenience and expense. The second options is a low-dose treatment with medications that are normally used to treat malaria-- hydroxychloroquine or chloroquine. This treatment maybe more convenient and cost-effective but it is not option prescribed as primary treatment. This study compares these two treatments.
The research questions are:
Is a low-dose regimen of hydroxychloroquine effective as phlebotomies for PCT treatment?
Are people treated with low-dose hydroxychloroquine more likely to have relapses of their PCT?
About this Study
This study compares two treatments for PCT. The first phase of the study is when patients get the treatment, either phlebotomies or hydroxychloroquine. During this phase study visits are every 2-4 weeks. After the treatment phase when participants have reached remission participants will meet with their doctor every 3-6 months, for 3 years to see if any symptoms return. The procedures done at these visits include physical exams, standard lab work such as checking iron levels, and lab work to look for porphyrin levels in the blood and urine.
A total of 100 patients with well-documented PCT will be enrolled in this study.
For each visit, you will be asked to:
Keep a log of your study medication if you are treated with hydroxychloroquine
Fill out a questionnaire about your PCT symptoms
Give a blood sample
1 to 2 teaspoons if you are treated by hydroxychloroquine
1 unit (450 ml, which is approximately 1 pint) of blood at regular intervals if you are treated with phlebotomy.
Give a urine sample
Have your local doctors office send us medical records and lab results
Targeted Enrollment
To be eligible to participate, you must:
Be at least 18 years old
Have well-documented PCT
Be able to provide consent to participate after you are informed about what the study includes
Be willing to avoid becoming pregnant during treatment; if you are a woman capable of becoming pregnant, you must use an effective contraceptive method
You are not eligible to participate if you have:
Your skin problem is not due to PCT, or is due to another type of porphyria
You are pregnant or if you plan to become pregnant during treatment
You have had prior treatment by phlebotomy, hydroxychloroquine or chloroquine within one month of starting this study. The exception to this is if you have specific medical records and lab results from before and during the treatment
You are unwilling or unable to comply with study procedures and visit schedule
You have participated in this study before
The neurologist December 2006, Volume 77, Issue 12, pp 1501-1505 Cite as Acute intermittent porphyria A clinical chameleon: Case report of a 40-year-old female patient
Monday - June 18, 2018 @ 07:00:06
The neurologist
December 2006, Volume 77, Issue 12, pp 1501-1505 Cite as
Acute intermittent porphyria A clinical chameleon: Case report of a 40-year-old female patient Authors Authors and affiliations M. ZimmermannEmail authorC. BonaccursoC. ValeriusG.F. Hamann 1. Case reports 238 Downloads 3 Citations Summary Acute porphyrias, although rare, often lead to misdiagnosed and dramatic clinical pictures. Various clinical pictures can be mimicked by the combination of internal, psychiatric and neurological symptoms. The report is about a 40-year-old female patient presented with a subacute tetraparesis in our clinic. In the previous months, the patient had been treated several times for abdominal symptoms in a medical and later on because of delirious symptoms with hallucinations in a psychiatric hospital. The typical triad of motor axonal neuropathy, abdominal discomfort, and psychiatric symptoms quickly led to the diagnosis of acute intermittent porphyria by determining urinary metabolites of porphyrin. The prognosis of acute porphyrias has improved significantly with insights into the pathogenesis of these diseases. The determination of the enzyme activities of the porphyrin metabolism also enables a diagnosis at the symptom-free interval. In the case of a positive family history, molecular genetic mutation screening may identify asymptomatic family members at increased risk of developing porphyria.
keywords Acute porphyrias Axonal neuropathy Heme synthesis Molecular genetic mutation screening Acute intermittent porphyria A clinical chameleon: case study of a 40-year-old female patient Summary Acute porphyrias are rare, but often misdiagnosed and may take a dramatic clinical course. The combination of various internal, psychiatrical and neurological symptoms can mimic different other diseases. We report a 40-year-old female patient who was admitted with a subacute tetraparesis. During the last two months the patient was treated for acute mental changes and hallucinations. The typical combination of abdominal pain, motor neuropathy and psychiatric symptoms confirmed by increased amounts of porphyrins and their precursors, led us to promptly diagnose acute intermittent porphyria. Better knowledge about the pathogenesis has clearly improved the prognosis of acute porphyria. In remission, measurement of enzyme activities or mutation screening can only be performed. A mutation screening for family members should be conducted to identify symptom-free carriers, especially in cases of a positive family history.
Literature
1.
Anderson KE, Spitz IM, Bardin CW, Kappas A (1990) A gonadotropin releasing hormone analogue prevents cyclical attacks of porphyria. Arch Intern Med 150: 14691474CrossRefPubMedGoogle Scholar
2.
Crimlisk HL (1997) The little imitator-porphyria: a neuropsychiatric disorder. J Neurol Neurosurg Psychiatry 62: 319328PubMedGoogle Scholar
3.
Dichter HN, Taddeini L, Lin S, Ayala GF (1977) Delta amino levulinic acid. Effect of a porphyrin precursor on an isolated neuronal preparation. Brain Res 126: 189195CrossRefPubMedGoogle Scholar
4.
Doss MO, Kuhnel A, Gross U (2000) Alcohol and porphyrin metabolism. Alcohol Alcohol 35: 109125PubMedGoogle Scholar
Gross U, Honcamp M, Daume E et al. (1995) Hormonal oral contraceptives, urinary porphyrin excretion and porphyrias. Horm Metab Res 27(8): 379383PubMedGoogle Scholar
Lindberg RL, Martini R, Baumgartner M et al. (1999) Motor neuropathy in porphobilinogen deaminase-deficient mice imitates the peripheral neuropathy of human acute porphyria. J Clin Invest 103(8): 11271134PubMedGoogle Scholar
10.
Meyer UA, Schuurmans MM, Lindberg RL (1998) Acute porphyrias: pathogenesis of neurological manifestations. Semin Liver Dis 18(1): 4352PubMedGoogle Scholar
11.
Pischik E, Bulyanitsa A, Kazakov V, Kaupinnen R (2004) Clinical features predictive of a poor prognosis in acute porphyria. J Neurol 251(12): 15381541CrossRefPubMedGoogle Scholar
12.
Sommer N, Gleichmann M (2003) Polyneuropathien. In: Brandt T, Dichgans J, Diener HC (Hrsg) Therapie und Verlauf neurologischer Erkrankungen, 4. Aufl. Kohlhammer, Stuttgart, S 12031217Google Scholar
13.
Soonawalla ZF, Orug T, Badminton MN et al. (2004) Liver transplantation as a cure for acute intermittent porphyria. Lancet 363: 705706CrossRefPubMedGoogle Scholar
14.
Yen PS; Chen CJ, Lui CC et al. (2002) Diffusion-weighted magnetic imaging of porphyric encephalopathy: a case report. Eur Neurol 48: 119121CrossRefPubMedoogle Scholar
STUDY 7204 GET INVOLVED STUDIES 7204 Clinical Diagnosis of Acute Porphyria
In order to participate in a study, you must personally contact Edrin @ the APF 1/866/APF/3635
Status: Recruiting
For Diseases:
Acute Intermittent Porphyria (AIP)
Hereditary Coproporphyria (HCP)
Variegate Porphyria (VP)
Background
The porphyrias are a group of genetic diseases caused by disturbances in the formation of heme, an essential component of hemoglobin and other proteins, leading to either acute cutaneous symptoms. Acute porphyria is often difficult to diagnose because the symptoms of abdominal pain, nausea, vomiting, etc. are not specific to porphyria. The purpose of this study is to test whether a focused questionnaire and laboratory evaluation tool can better define risk factors associated with possible genetic porphyria.
The goals of this study are:
To determine whether first degree family members of patients with genetically-confirmed acute porphyria have symptoms and laboratory findings.
To devise a Genetic Carrier Profile that could be used to screen people in whom the diagnosis of porphyria is being considered.
To explain other possible causes of minor increases in porphyrin levels in patients with recurrent abdominal pain who have not been diagnosed with porphyria
About this Study
This is a longitudinal study will consist of two parts.
Part 1: 100 people. We will enroll individuals who are first-degree relatives (child, sibling, or parent) of a patient with a genetically-confirmed diagnosis of one of the acute porphyrias (called the index case). We are interested in all three types of acute porphyria: Acute Intermittent Porphyria (AIP), Hereditary Corproporphyria (HCP) or Variegate Porphyria (VP). Participants (the first-degree relatives) must not have had any genetic testing in the past. They will have a study visit during which they will complete a history questionnaire and have routine laboratory tests, including genetic testing for the acute porphyria mutation that is in their family. The researchers will use this data to develop a Clinical Profile of the risk factors associated with being a genetic carrier of acute porphyria.
Part 2:Not Yet Recruiting Researchers will test the validity of the Clinical Profile. This part of the study will include subjects who are thought to possibly have acute porphyria, but do not have a confirmed.
Participation in Parts 1 and 2 of the study include one study visit with the following:
A physical exam
Answering questions about your health and quality of life
Donation of Giving samples for laboratory evaluations, including samples of blood, urine, and possibly stool
Donation of Giving a blood sample for genetic testing
Review of your medical records
Targeted Enrollment
To be eligible to participate in Part 1, you must:
Be 15 years of age or older
Be a first-degree relative (child, sibling, or parent) of an individual with genetically proven acute porphyria (AIP, HCP or VP)
Not have had any previous genetic testing for acute porphyria
To be eligible to participate in Part 2, you must:
Be 15 years of age or older
Have a history of suggestive clinical features, such as abdominal, back or limb pain, recurrent nausea lasting days, reaction to medications, etc.
An increase in urinary, fecal or serum porphobilinogen (PBG) and/or porphyrins
How to participate:
In order to participate in a study, you must personally contact Edrin @ the APF 1/866/APF/3635
OVERVIEW: What every practitioner needs to know Porphyrias are a very rare group of metabolic disorders.
Porphyrias are a very rare group of metabolic disorders.
The disease is a consequence of defects in the normal metabolic pathway for the biosynthesis of heme, with accumulation of porphyrins, which are the intermediates in the pathway of heme synthesis.
The term Porphyria comes from the Greek word porphyra, which means purple.
Are you sure your patient has Porphyria? What are the typical findings for this disease?
There are two major types of Porphyrias, hepatic or erythropoietic, depending on whether the intermediates that accumulate come from the liver or from developing erythrocytes.
The porphyrias can also be classified based on the clinical manifestations of:
-Acute Porphyrias, with severe bouts of abdominal pain that are recurrent.
-Cutaneous Porphyrias, presenting with painful skin lesions.
The main clinical manifestations are cutaneous photosensitivity with painful skin lesions and abdominal pain as a result of a neurologic dysfunction.
-The liver plays two major roles in the metabolism of the porphyrins. It is the major site of porphyrin production as part of heme synthesis. The liver is also important in the excretion of the more lipophilic porphyrins. For example, protoporphyrin is only excreted in bile.
See Figure 1 for a diagram depicting heme synthesis, and Table I for the classification, clinical and laboratory features of the porphyrias .
Of the five hepatic porphyrias, ADP, AIP, HCP, and VP present with acute neurological attacks (abdominal pain, peripheral neuropathy) and elevated levels of one or more of the porphyrin precursors aminolevulinic acid (ALA) and porphobilinogen (PBG); these are the acute porphyrias. PCT usually presents in adults with blistering lesions in the skin but not acute attacks. It is important to know that HCP and VP also could have cutaneous manifestations.
The erythropoietic porphyrias, CEP and EPP, usually present with elevation of porphyrins in the bone marrow and erythrocytes. They present with cutaneous photosensitivity in infancy. Manifestations of CEP are similar to those of PCT. The lesions in EPP are more painful, and non-blistering. EPP causes symptoms before puberty.
ALA dehydratase deficiency porphyria (ADP)
Genetics: ADP is a rare autosomal recessive acute hepatic porphyria secondary to the deficiency of ALA dehydratase activity.
Incidence: There are only six documented cases, with five in children or adolescents with specific gene mutations identified.
Etiology:The affected homozygotes have less than 10% of the normal ALA dehydratase enzyme activity in the erythrocytes.
Frequency: The frequency is unknown, but the frequency of the heterozygotes with less than 50% of the enzyme activity was approximately 2% in a population study from Sweden.
Clinical features: The presentation is variable and depends of the amount of enzyme activity. All the patients had marked decrease of the ALA dehydratase activity. Four of the patients were adolescents presenting with abdominal pain and neuropathy, similar to the symptoms seen in AIP. There was one infant with more severe symptoms presenting with failure to thrive early after birth.
Diagnosis: Patients have increased levels of urinary ALA and COPRO III. The urinary PBG is slightly elevated or normal. ALA dehydratase activity in the erythrocytes is less than 10% than the normal level. Homozygotes have less than 10% of the enzyme activity.
Differential diagnosis: Tyrosinemia type 1 and lead intoxication can inhibit ALA dehydratase, increase urinary ALA, and cause symptoms similar to those seen during the acute attacks.
Treatment: The four males that developed attacks during adolescence were treated with intravenous hemin, as in AIP. The infant that presented with more severe symptoms and did not survive, did not respond to hemin or liver transplantation; he received supportive therapy with blood transfusions and total parenteral nutrition.
Acute intermittent porphyria (AIP)
Genetics: This hepatic porphyria is an autosomal dominant disorder as a result of having half the normal activity of hydroxymethylbilane (HMB)-synthase. In heterozygous individuals, the presentation is variable.
Incidence: The disease is widespread, but the actual incidence is unknown. It is more common in Scandinavia and Great Britain.
Pathophysiology: The disease is often activated by different environmental or hormonal factors such as drugs, diet, and steroid hormones in individuals with the genetic predisposition. Once the precipitating factor is known, the attacks can be prevented by avoiding triggers.
Clinical features:AIP remains latent in the vast majority of patients with the mutation, especially before puberty. Apparently, having half the activity of the HMB-synthase enzyme is sufficient for normal hepatic heme synthesis. Also the induction of ALAS1 is thought to underlie acute attacks in AIP. When heme synthesis is increased in the liver, half-normal HMB-synthase may become limiting and ALA, PBG and other heme pathway intermediates may accumulate.
The precipitating factors include endogenous and exogenous gonadal steroids, porphyrinogenic drugs, alcohol ingestion, and a low calorie diet. Symptoms are more common in females and after puberty, suggesting that hormones play an important role in clinical expression. Pregnancy is usually well tolerated. An extensive list of safe and unsafe drugs for individuals with porphyria is provided at The Drug Database for Acute Porphyria, www.drugs-porphyria.com. Attacks can be provoked by surgery, infections, and alcohol, but many patients with recurrent attacks cannot identify an instigating factor. An increase in carbohydrates may ameliorate the attacks. The disease is rarely fatal if identified promptly.
Abdominal pain is the most common symptom; it usually is steady, dull, and poorly localized. Patients often have constipation, abdominal distension, and decreased bowel sounds. Inflammation is absent; therefore, fever and leukocytosis are not prominent. Other manifestations include nausea; vomiting; tachycardia; hypertension; extremity, neck and chest pain; headaches; muscle weakness; sensory loss; tremors; sweating; dysuria; and bladder distension. Seizures could be a neurologic manifestation of the disease but could also be secondary to hyponatremia. Long-term risks for hypertension, impaired renal function. and hepatocellular carcinoma are increased.
Low grade abnormalities in the liver function tests are common. The risk of hepatocellular carcinoma is increased.
Diagnosis:ALA and PBG are increased in plasma and urine during the acute attacks. The diagnosis of an acute attack is mostly based on the clinical findings. The levels of the porphyrin precursors decrease after an attack but usually remain elevated. Dramatic decreases of the porphyrin precursors are seen after treatment with hemin. A normal urinary PBG excludes AIP as a cause of the current symptoms. Unlike HCP and VP, fecal and plasma porphyrins are normally or minimally elevated in AIP. The deficient enzyme is detectable in the erythrocytes of most AIP heterozygotes. Any condition that increases erythropoiesis or younger erythrocytes will present levels of enzyme activity in the normal ranges. More than 240 HMB-synthase mutations have been identified in AIP. Yearly hepatic imaging to assess for hepatocellular carcinoma is recommended. In these patients, there is no elevation of α-fetoprotein.
Treatment: Narcotic analgesics are used for the abdominal pain seen during the attacks. Phenothiazines are useful to treat nausea, vomiting, anxiety, and restlessness. Intravenous glucose (at least 300 g/dl) is helpful in milder attacks. Hemin is very effective and should be started immediately for severe attacks and for milder attacks that do not respond to glucose infusion within 1-2 days. It is used as 3-4 mg of heme as lyophilized hematin, heme albumin, or heme arginate infused daily for 4 days. Recovery is usually rapid if treatment is started early. Patients with neuropathy could require months to recover. One teenage patient that suffered many attacks responded well to an orthotopic liver transplantation, with normalization of urinary levels of PBG and ALA, but liver transplantation should not be considered as first-line treatment in AIP.
Homozygous dominant AIP:This is a very rare form of porphyria. It presents in infancy. Patients have extremely low HMB-synthase (<2%) activity. This condition has been described in 4 European children from different families, except for two siblings. All patients presented with failure to thrive, developmental delay, cataracts, and/or hepatosplenomegaly, but with no acute attacks.
Porphyria cutanea tarda (PCT)
Genetics: Hepatic URO-decarboxylase is deficient in all types of PCT. About 80% of the patients have no mutation of the enzyme and are said to have type 1 disease, or type 3 if relatives are affected. Patients with heterozygous mutation for the enzyme have type 2 disease (familial PCT). It is important to know that having decreased enzyme activity per seis not sufficient to develop symptoms; patients also need other genetic and environmental factors.
Incidence: This is the most common porphyria. It could be sporadic (type 1) or familial (types 2 and 3). It is also seen after exposure to halogenated aromatic hydrocarbons.
Clinical features: Deficient hepatic uroporphyrinogen (URO)-decarboxylase is present in all types of PCT, and should be about or less than 20% of the normal activity. It is believed that there is a specific inhibitor of the enzyme in the liver acting in the presence of iron and under conditions of oxidative stress. There is increased prevalence of PCT type 1 and 2 in patients with the common hemochromatosis mutations. PCT is strongly associated with hepatitis C, excess alcohol ingestion, and estrogen use, as well as human immunodeficiency virus. Multiple factors that appear to act synergistically can be identified in the individual patient with PCT. The major clinical feature is blistering lesions on the back of the hands, and also on the forearms, face, legs and feet. There is hypertrichosis, hyperpigmentation, and skin friability with small white papules (milia). Sun-exposed skin usually becomes thickened. Patients have evidence of chronic liver disease with abnormal liver function tests. Cirrhosis and hepatocellular carcinoma may develop in the long term.
Diagnosis: Porphyrins are increased in the liver, plasma, urine, and stools. The urinary ALA is mildly elevated, but the PBG level is normal. Plasma porphyrins are also increased, which is useful for screening. There is an increase of isocoproporphyrins, especially in feces; this is diagnostic for URO-decarboxylase deficiency. This enzyme activity in the erythrocytes is about half of the normal level in type PCT and normal in type 1 and type 3. More than 65 mutations have been identified in the URO-decarboxylase gene in type 2 PCT and hepatoerythropoietic porphyria (HEP).
Treatment: Discontinuation of the risk factors (alcohol, estrogens, and iron supplements) is the first step in treatment, but it could take time for the patient's condition to respond. A complete resolution can almost be achieved with repeated phlebotomy to reduce the hepatic iron. A unit of blood can be removed every two weeks until the ferritin level is at the lower limit of normal. In order to document improvement in PCT, the total plasma porphyrin concentration must be followed; its value returns to normal after the target level of ferritin is reached. The plasma porphyrin levels must be followed every 6-12 months for early detection of recurrences. If the patient does not tolerate phlebotomy, an alternative is to start a low-dose regimen with chloroquine (125 mg twice weekly) or hydroxychloroquine. Treatment of patients with concurrent hepatitis C should be postponed until after the PCT is treated and in remission.
Hepatoerythropoietic porphyria (HEP): This is the homozygous form of type 2 PCT. Most patients have inherited mutations from unrelated parents. In HEP, the enzyme activity is approximately 3%-10% of normal. Excess porphyrins originate from the liver. Patients usually have blistering skin lesions, hypertrichosis, scarring, and red urine during infancy or childhood. Concurrent conditions that affect the liver may worsen the disease severity; this has been described in a toddler with hepatitis A. Avoidance of sunlight is very important. The outcome depends on the enzyme activity, and it is better if sunlight can be avoided. The use of phlebotomy has shown no benefit.
Hereditary coproporphyria (HCP)
Genetics: HCP is an autosomal dominant hepatic porphyria resulting from half the normal activity of the enzyme COPRO-oxidase.
Incidence: This porphyria is less common than AIP and VP. It is more common in women.
Clinical features: The disease presents with very similar symptoms to AIP. The attacks are less severe than those in AIP. Cutaneous photosensitivity may occur, but less commonly than in VP. HCP is influenced by the same factors as AIP. The disease is latent before puberty. Although HCP is less severe than AIP, fatal motor neuropathy may nevertheless occur. Blistering skin lesions are identical to those in PCT and VP, and begin in childhood in rare homozygous cases.
Diagnosis: The diagnosis is confirmed by increased fecal coproporphyrin III; this is also elevated in urine. It will remain elevated in feces even if there are no symptoms.
Treatment: The neurologic symptoms are treated as in AIP. Phlebotomy and chloroquine are ineffective in treating the cutaneous manifestations.
Variegate porphyria (VP)
Genetics: VP is an autosomal dominant hepatic porphyria. It is secondary to deficient PROTO-oxidase enzyme activity.
Incidence: This disease is very common in South Africa, where 3 of every 1000 whites have the disorder. In other countries, VP is less common than AIP.
Clinical features: The attacks are usually triggered by diet, drugs, or hormones. They are identical to the attacks seen in AIP, but milder and less often fatal. The skin lesions are the same as those in PCT, but usually more difficult to treat. In two large studies of VP, 59% of study subjects had only skin lesions, 20% had only acute attacks, and 22% had both.
Diagnosis: ALA and PBG are elevated in urine during the attacks. Coproporphyrin III is elevated in urine and feces. The fluorescence emission spectrum of porphyrins in plasma at a neutral pH in VP is distinctive and rules out other porphyrias, especially PCT, which is common.
Treatment: During acute attacks, the treatment is the same as in AIP. Hemin should be started early. There are few effective methods to prevent skin lesions, but sun exposure should be avoided and protective clothing should be worn. Phlebotomy, β-carotene and chloroquine are ineffective.
Congenital erythropoietic porphyria (CEP)
Genetics:CEP is an autosomal recessive disorder, and is also known as Gunther disease. It is secondary to a marked deficiency in the activity of URO-synthase, with accumulation of uroporphyrin I and coproporphyrin I isomers.
Clinical features: The symptoms start early in infancy, with severe cutaneous photosensitivity. It could cause nonimmune hydrops fetalis. The exposed skin is friable, with bullae and vesicles that are prone to rupture and infection. Skin thickening, focal hypo- or hyperpigmentation, and hypertrichosis of the face and extremities are characteristic of CEP. Skin infection and bone resorption can lead to disfigurement of the face and hands. The teeth are reddish-brown and fluorescent on exposure to long-wave ultraviolet light, due to the deposition of excess porphyrins. Patients present with hemolysis leading to splenomegaly, also due to increased deposition in the red blood cells.
Diagnosis: Uroporphyrin I and coproporphyrin I accumulate in the bone marrow, red blood cells, plasma, urine, and feces. The confirmation of the diagnosis is made by the identification of the specific mutations in the URO-synthase gene or by the decreased activity of the enzyme.
Treatment: Transfusion for anemia is required in severe cases, even in utero. Recurrent transfusions are effective to suppress erythropoiesis, but result in iron overload and other complications. Splenectomy may reduce hemolysis and decrease transfusion requirements. Protection from sunlight is essential, and minor skin trauma should be avoided. Recently, bone marrow and cord blood transplantation have proven effective in several transfusion-dependent children.
Erythropoietic protoporhyria (EPP)
Genetics: EPP is an autosomal dominant inherited disorder resulting from partial deficiency of ferrochelatase (FECH), approximately 15%-25% of the enzyme activity. A polymorphism in intron 3 of the wild-type allele affects the level of enzyme activity and clinical expression.
Incidence: It is the most common porphyria in children and the second most common in adults.
Pathophysiology: Protoporphyrin accumulates in the bone marrow, then appears in plasma, is taken up in the liver, and is excreted in bile and feces. It is also transported to the skin, causing photosensitivity.
Clinical features: Skin photosensitivity starts in childhood. The skin becomes painful, erythematous, and itchy very soon after sun exposure, similar to angioedema. Vesicular lesions are uncommon. Vesicles and bullae occur in only 10% of cases. Chronic lesions are lichenification, labial grooving, leathery pseudovesicles, and nail changes. The skin photosensitivity remains stable over many years in most patients. There are no other factors that exacerbate EPP, as seen in hepatic porphyrias. Up to 20% of the patients have minor liver function abnormalities. It has been reported that 5% of the cases have rapid progression of liver disease, with acute liver failure and death. This liver disease may cause abdominal pain, especially in the RUQ, and back pain.
Diagnosis: An increase of the free erythrocyte protoporphyrin, not complexed with zinc, is the hallmark of the disease. Protoporphyrin levels are also elevated in bone marrow, plasma, bile, and feces. Plasma and fecal porphyrins are less increased or sometimes normal; this is in contrast to other cutaneous porphyrias. Erythrocyte protoporphyrin concentrations are increased lead poisoning, iron deficiency, various hemolytic disorders and all homozygous forms of porphyria, but in all these other conditions is always complexed with zinc.
Treatment: Avoiding sunlight exposure and wearing appropriate clothing are essential. Oral �-carotene (120-180 mg/dL) improves tolerance to sunlight in many patients if the dose is adjusted to maintain a serum carotene level in the range of 10-15 µmol/L (600-800 µg/dL). Oral �-carotene causes mild skin decoloration due to carotenemia. Treatment of the hepatic complications is difficult. Cholestyramine and activated charcoal may interrupt the enterohepatic circulation of protoporphyrin and promote its fecal excretion, leading to some improvement. Plasmapheresis and intravenous hemin are sometimes helpful. Splenectomy may be indicated in the presence of splenomegaly and hemolysis. Liver transplantation is sometimes necessary and often successful in the short term; however, EEP often recurs in the transplanted liver due to reaccumulation of protoporphyrin. Treatment with hemin and plasmapheresis may prevent or delay the reaccumulation. Bone marrow transplantation has been successful in humans with EPP, this should be considered after liver transplantation if a suitable donor can be found.
What other disease/condition shares some of these symptoms?
It is always important to rule out a more common etiology for abdominal pain since many patients present with recurrent abdominal pain.
Patients with chronic liver disease could have some similar findings in the urine analysis of the porphyrins.
Tyrosinemia type 1 and lead intoxication should be ruled out in patients presenting with symptoms similar to those seen in ADP.
What caused this disease to develop at this time?
The accumulation of metabolic intermediates in these patients is the reason why the disease develops and symptoms start. The elucidation of the pathway of the heme synthesis clarified the abnormal accumulation of metabolic intermediates in patients with porphyrias.
Knowing the properties of the porphyrins is important in order to understand the clinical manifestations of the disease.
There is an excessive accumulation and excretion of porphyrins and their precursors as a result of specific enzymatic defects in the heme synthetic pathway.
The condition could be inherited or acquired. There are many patients with no clinical manifestations who have the enzymatic defect.
There are several precipitating factors for an acute and potentially fatal attack, such as drugs, alcohol, fasting, or stress. This is the reason why it is very important to try to identify these patients and prevent an attack.
A rapid screening of porphobilinogen (PBG) is very useful when a patient presents with a suspected visceral attack.
In patients with suspected cutaneous porphyria, screening tests for increased erythrocytic porphyrins should be performed, as well as screening tests for urinary porphyrins.
What laboratory studies should you request to help confirm the diagnosis? How should you interpret the results?
Positive screening tests always must be confirmed with specific quantitative tests.
Porphyrins have a unique photo optic property that allows them to absorb light in the long ultraviolet range and emit an intense fluorescence. This is very useful in detecting porphyrins in biologic specimens and forms the basis of many diagnostic screening tests for the porphyrias.
DNA-based tests as well as enzymatic assays are helpful for evaluation, genetic diagnosis, and identifying the particular defect. But these are not useful when trying to provide a rapid diagnosis in a symptomatic patient.
Would imaging studies be helpful? If so, which ones?
Some patients with different types of porphyrias like AIP and ADP have an increased incidence of hepatocellular carcinoma (HCC). For these patients, an imaging study of the abdomen (ultrasound or CT scan) is necessary, especially during adulthood.
Confirming the diagnosis
An acute porphyria should be suspected in patients with recurrent neurovisceral symptoms after puberty, such as abdominal pain. If an initial evaluation does not yield any other diagnosis, then urinary ALA and PBG should be checked.
Urinary PBG is almost always elevated during an acute attack in AIP, HCP, and VP.
A spot urine test for PBG could be very helpful in obtaining a diagnosis of porphyria. A 24-hour collection may delay the diagnosis.
Patients with chronic liver disease could have elevation of urinary porphyrins; therefore, measurements of the porphyrins alone should be avoided.
Blistering skin lesions are almost always accompanied by elevation of total plasmaporphyrins.
Confirmation at the DNA level by identifying the mutation or mutations is important after the diagnosis is established, and permits family studies.
If you are able to confirm that the patient has a porphyria, what treatment should be initiated?
The treatment will always depend on the type of porphyria (see above for specific treatments in each different type of the disease).
What causes this disease and how frequent is it?
See above for each type of porphyria.
What complications might you expect from the disease or treatment of the disease?
The increased predisposition for hepatocellular carcinoma is seen in various types of porphyria.
Evaluation of the patient with abdominal pain and suspected porphyria
Porphyria should suspected in the pediatric population, particularly in adolescents who present with abdominal pain and no obvious clinical explanation.
The symptoms are very similar to the ones seen in functional abdominal pain, poorly localized pain, no rebound tenderness, constipation, and psychological instability. Due to these non-specific symptoms, a high index of suspicion is necessary for the diagnosis of porphyria.
Patients with true acute porphyria excrete increased amounts of urinary ALA and usually PBG. The absence of these during an acute attack rules out the diagnosis of porphyria.
An incorrect diagnosis of porphyria could be made in patients with functional abdominal pain with the finding of elevated total porphyrins in urine, which is a laboratory abnormality commonly found in secondary porphyrinuria.
Measurement of fecal porphyrins is also helpful in discriminating HCP and VP in remission from patients with secondary porphyrinurias. Fecal coproporphyrin is markedly elevated in these porphyrias, whereas it is normal in patients with secondary porphyrinuria.
How can Porphyrias be prevented?
Prevention is the central component in the management of patients with this condition.
The use of high carbohydrate intake, intravenous hematin, and pain control are crucial in the treatment of acute visceral attacks.
Sun avoidance and strong skin protection are very important in reducing cutaneous manifestations and complications.
Avoidance of certain drugs, surgery, certain diets, and steroid hormones are of crucial importance.
What is the evidence?
As described above, there are different treatments for the different types of porphyrias. The evidence is difficult to gather since this is a very rare condition, but treatments such as hemin and intravenous glucose infusion in AIP have been shown to be effective in these patients. It is very important to avoid triggers of the attacks, such as certain drugs.
The following references are recommended for further information in porphyrias:
-Classification of the Porphyrias
Anderson, KE, Bloomer, JR, Bonkovsky, HL. "Recommendations for the diagnosis and treatment of the acute porphyrias". Ann Intern Med. vol. 142. 2005. pp. 439-50.
Kauppinen, R. "Porphyrias". Lancet. vol. 365. 2005. pp. 241-52.
-ALA dehydratase deficiency porphyria (ADP)
Shady, AA, Colby, BR, Cunha, LF. "Congenital erythropoietic porphyria: identification and expression of eight novel mutations in the uroporphyrinogen III synthase gene". Br J Haematol. vol. 117. 2002. pp. 980-7.
Anderson, KE, Bloomer, JR, Bonkovsky, HL. "Recommendations for the diagnosis and treatment of the acute porphyrias". Ann Intern Med. vol. 142. 2005. pp. 439-50.
Sassa, S. "ALAD porphyria". Semin Liver Dis. vol. 18. 1998. pp. 95-101.
Doss, MO, Stauch, T, Gross, U. "The third case of Doss porphyria (delta-amino-levulinic acid dehydratase deficiency) in Germany". J Inherit Metab Dis. vol. 27. 2004. pp. 529-36.
Akagi, R, Kato, N, Inoue, R. "delta-Aminolevulinate dehydratase (ALAD) porphyria: the first case in North America with two novel ALAD mutations". Mol Genet Metab. vol. 87. 2006. pp. 329-36.
-Acute intermittent porphyria (AIP)
Handschin, C, Lin, J, Rhee, J. "Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1alpha". Cell. vol. 122. 2005. pp. 505-15.
Anderson, KE, Bloomer, JR, Bonkovsky, HL. "Recommendations for the diagnosis and treatment of the acute porphyrias". Ann Intern Med. vol. 142. 2005. pp. 439-50.
Soonawalla, ZF, Orug, T, Badminton, MN. "Liver transplantation as a cure for acute intermittent porphyria". Lancet. vol. 363. 2004. pp. 705-6.
Bonkovsky, HL, Poh-Fitzpatrick, M, Pimstone, N. "Porphyria cutanea tarda, hepatitis C, and HFE gene mutations in North America". Hepatology. vol. 27. 1998. pp. 1661-9.
Mehrany, K, Drage, LA, Brandhagen, DJ. "Association of porphyria cutanea tarda with hereditary hemochromatosis". J Am Acad Dermatol. vol. 51. 2004. pp. 205-11.
Egger, NG, Goeger, DE, Payne, DA. "Porphyria cutanea tarda: multiplicity of risk factors including HFE mutations, hepatitis C, and inherited uroporphyrinogen decarboxylase deficiency". Dig Dis Sci. vol. 47. 2002. pp. 419-26.
Bonkovsky, HL, Barnard, GF. "Diagnosis of porphyric syndromes: a practical approach in the era of molecular biology". Semin Liver Dis. vol. 18. 1998. pp. 57-65.
Thunell, S, Harper, P. "Porphyrins, porphyrin metabolism, porphyrias. III. Diagnosis, care and monitoring in porphyria cutanea tarda--suggestions for a handling programme".Scand J Clin Lab Invest. vol. 60. 2000. pp. 561-79.
-Hereditary coproporphyria (HCP)
Martásek, P. "Hereditary coproporphyria". Semin Liver Dis. vol. 18. 1998. pp. 25-32.
Anderson, KE, Bloomer, JR, Bonkovsky, HL. "Recommendations for the diagnosis and treatment of the acute porphyrias". Ann Intern Med. vol. 142. 2005. pp. 439-50.
-Variegate porphyria (VP)
Schmitt, C, Gouya, L, Malonova, E. "Mutations in human CPO gene predict clinical expression of either hepatic hereditary coproporphyria or erythropoietic harderoporphyria". Hum Mol Genet. vol. 14. 2005. pp. 3089-98.
Hift, RJ, Meissner, PN, Corrigall, AV. "Variegate porphyria in South Africa, 1688-1996--new developments in an old disease". S Afr Med J. vol. 87. 1997. pp. 722-31.
Bonkovsky, HL, Barnard, GF. "Diagnosis of porphyric syndromes: a practical approach in the era of molecular biology". Semin Liver Dis. vol. 18. 1998. pp. 57-65.
Poh-Fitzpatrick, MB. "A plasma porphyrin fluorescence marker for variegate porphyria".Arch Dermatol. vol. 116. 1980. pp. 543-7.
-Congenital erythropoietic porphyria (CEP)
Piomelli, S, Poh-Fitzpatrick, MB, Seaman, C. "Complete suppression of the symptoms of congenital erythropoietic porphyria by long-term treatment wtih high-level transfusions".N Engl J Med. vol. 314. 1986. pp. 1029-31.
Davenport, C, Dillon, WP, Sze, G. "Neuroradiology of the immunosuppressed state".Radiol Clin North Am. vol. 30. 1992. pp. 611-37.
Shaw, PH, Mancini, AJ, McConnell, JP. "Treatment of congenital erythropoietic porphyria in children by allogeneic stem cell transplantation: a case report and review of the literature". Bone Marrow Transplant. vol. 27. 2001. pp. 101-5.
Rank, JM, Carithers, R, Bloomer, J. "Evidence for neurological dysfunction in end-stage protoporphyric liver disease". Hepatology. vol. 18. 1993. pp. 1404-9.
Poh-Fitzpatrick, MB, Wang, X, Anderson, KE. "Erythropoietic protoporphyria: altered phenotype after bone marrow transplantation for myelogenous leukemia in a patient heteroallelic for ferrochelatase gene mutations". J Am Acad Dermatol. vol. 46. 202. pp. 861-6.
-Other important references
Chemmanur, AT, Bonkovsky, HL. "Hepatic porphyrias: diagnosis and management". Clin Liver Dis. vol. 8. 2004. pp. 807-38.
Bloomer, JR, Bonkovsky, HL. "The porphyrias". Dis Mon. vol. 35. 1989. pp. 1-54.
Morton, KO, Schneider, F, Weimer, MK. "Hepatic and bile porphyrins in patients with protoporphyria and liver failure". Gastroenterology. vol. 94. 1988. pp. 1488-92.
Bonkovsky, HL, Barnard, GF. "Diagnosis of porphyric syndromes: a practical approach in the era of molecular biology". Semin Liver Dis. vol. 18. 1998. pp. 57-65.
No sponsor or advertiser has participated in, approved or paid for the content provided by Decision Support in Medicine LLC. The Licensed Content is the property of and copyrighted by DSM.
The porphyrias are a group of rare metabolic diseases that may present in childhood or adult life and are due to deficiencies of enzymes in the heme biosynthetic pathway. Porphyrias have various symptoms depending on the type, but these can range from neurological symptoms to sun sensitivity. See the descriptions of each type to get more information. The natural history of these disorders is not well described and it is not known why some patients are more severe than others. Therefore, the purpose of this long-term follow-up study is to collect a large group of patients with the different types of porphyria and to provide a better understanding of the natural history of these disorders. The hope is that this information will help in developing new forms of treatment.
The research aims are:
To study the prevalence of specific indicators of disease severity. To study the effects on quality of life and health of various porphyrias.
To determine the relationships between disease severity and various biological characteristics, genetic information, and environmental factors.
About this Study
This is an observational, longitudinal study of approximately 800 individuals with the various types of porphyria. Those participating will be evaluated annually for 5 years, or longer if they agree.
Participation in this project will include:
Participating in annual visits or contacts.
Providing samples, including blood, buccal cells (cells from inside of the mouth), saliva, and urine. The type of samples and amounts may vary from person to person, depending on the type of porphyria.
Giving permission for samples to be stored and used for porphyrias research.
Granting permission to obtain your medical records.
Providing information about your medical history and family history.
Completing a questionnaire about your porphyria symptoms and quality of life.
Targeted Enrollment
To be eligible to participate, you must:
Have a confirmed diagnosis of one of the porphyrias (EPP, XLP, CEP, VP, AIP, HCP, PCT, HEP, ADP)
You are not eligible to participate if you have:
Elevations of porphyrins due to other diseases, such as liver and bone marrow diseases.
A prior diagnosis of porphyria that cannot be documented by existing medical records or repeat biochemical or DNA testing.
How to participate:
In order to participate in a study, you must personally contact the study coordinator of the participating institution closest to you by phone or e-mail. Please use the information below to inquire about participation.
University of California at San Francisco Yuvraaj Kapoor Phone: 415-476-8405 E-mail: Yuvraaj.kapoor@ucsf.edu
Florida
University of Miami Miller School of Medicine, Miami Odalys Rodriguez Bravo, RN, Research Coordinator Phone: (305) 243-4648 E-mail: obravo@med.miami.edu
New York
Ichan School of Medicine at Mount Sinai, New York Hetanshi Naik, Coordinator 212-241-7699 E-mail: Hetanshi.naik@mssm.edu
North Carolina
Wake Forest School of Medicine, Winston-Salem Dee Faust, Coordinator 336-713-1442 E-mail: delannin@wakehealth.edu
Texas
University of Texas Medical Branch, Galveston Csilla Hallberg, MD, Coordinator 409-772-4661 E-mail: porphyria.center@utmb.edu
Utah
University of Utah, Salt Lake City Sharada Dixit, Study Coordinator 801-587-7525 E-mail: Sharada.Dixit@hsc.utah.edu
Washington
University of Washington School of Medicine, Seattle Andrew Herstein, MD, Research Coordinator 206-288-7273 E-mail: aherstei@seattlecca.org
Diagnostic Recommendations PART 3
Friday - June 8, 2018 @ 07:00:13
DIAGNOSTIC RECOMMENDATIONS Initial diagnosis: The two most important diagnostic recommendations are to 1) maintain a high index of suspicion, and 2) be aware that laboratory testing is available that can readily make a diagnosis of acute porphyria or rule it out.
"Think porphyria" in patients with abdominal pain that is unexplained after an initial workup has excluded common causes such as appendicitis, cholecystitis, pancreatitis, etc. Associated findings such as reddish urine, tachycardia, hypertension, hyponatremia and proximal muscle weakness can be further indications for testing for porphyria. Other clues include use of potentially porphyrinogenic drugs such as sulfonamides, barbiturates, rifampin or metoclopramide, and premenstrual symptoms in women.
When acute porphyria is suspected, confirmation should be initiated immediately by biochemical testing. Measuring urinary PBG is most important for diagnosis of acute porphyrias. Porphobilinogen (PBG) excretion is normally 0-4 mg/day, and is approximately in the same range when expressed as mg/g creatinine or even as mg/L. In an acute attack, spot urine porphobilinogen (PBG) levels are substantially increased (20-200 mg/L).
The test should be established at the local hospital pathology clinical chemistry department. It is recommended that all medical centers provide for rapid testing for increased porphobilinogen(PBG), since sending samples out to a referral laboratory can greatly delay diagnosis and treatment. Thereby, when acute porphyria is suspected the diagnosis can be ruled in or out in a timely fashion.
If PBG is found to be elevated, treatment can be initiated without further delay. However, samples should be collected before treatment to establish the type of acute porphyria. This second-line testing should be comprehensive (Table below) - to include urinary ALA and porphyrins, plasma and fecal porphyrins, and erythrocyte porphobilinogen (PBG) deaminase, and does not require collection of 24 hour samples. DNA studies to identify the inherited mutation are important after the type of acute porphyria is established biochemically.
Recurrent attacks in a patient with proven acute porphyria are usually similar and can be diagnosed on clinical grounds, and without biochemical reconfirmation. Treatment may be initiated immediately after excluding other causes of the acute symptoms. However, the laboratory reports that were the basis for the prior diagnosis porphyria should be available for review to assure that the diagnosis was accurate.
The American Porphyria Foundation recommends known patients with hepatic porphyria wear medical alert bracelets or carry medical alert cards.
TREATMENT Panhematin Therapy The most effective therapy for the acute attack is Panhematin. This treatment is specific, because it corrects the deficiency of regulatory heme in the liver and down-regulates ALAS. Glucose loading has a similar effect, but is much less potent and effective and should be used only for mild attacks.
The standard Panhematin treatment course is 3-4 mg/kg by vein once daily for 4 days. If the diagnosis is confirmed, the first dose can be given in the ER. Panhematin (Recordati Rare Diseases) is the only hemin therapy available in the United States. It is a lyophilized preparation of hydroxyheme (hematin) and is FDA approved for treating acute porphyrias. To order Panhematin, contact Recordati Rare Diseases at: www.recordatirarediseases.com or 866-654-0539
Recordati Rare Diseases will express ship Panhematin to the pharmacy on request. Heme arginate is a hemin preparation available in Europe and South Africa and is administered at the same dose.
Panhematin can cause phlebitis at the site of infusion, which may compromise venous access. Experts recommend that lyophilized Panhematin be reconstituted with human albumin rather than sterile water, which enhances its stability and decreases the chance of phlebitis. The method for this has been published (5). In brief, the authors recommended reconstituting a 313 mg vial of Panhematin with 132 ml of 25% human serum albumin, then swirling 15-20 times to mix. The amount required is then withdrawn into a syringe using a 5-micron filter needle, and injected into a 150 ml sterile bottle prior to IV infusion. This must be placed in an amber bag to protect from light, and used within one hour of preparation, in part because the solution contains no bacteriostatic agents. The concentration of hemin as heme-albumin is 2.4 mg/mL, which is used to calculate the volume needed to deliver a dose of 3-4 mg/kg. The dose is generally infused by piggyback to an intravenous line that is infusing 0.9% sodium chloride at a moderate rate. The dose should be infused over a period of at least 60 minutes or at a rate that should not exceed 1 mL/min (5). Panhematin treatment can be continued if recovery is not evident within 4 days.
Complications of IV Panhematin therapy other than local phlebitis include fever, aching, malaise, and hemolysis. Rarely, there have been reports of transitory circulatory collapse and, after excessive dosing, renal failure. Panhematin has been given safely in pregnancy.
Patients should be monitored closely and treated for pain and other symptoms, and for complications such a respiratory impairment, hyponatremia and neuropsychiatric manifestions. Monitoring in an intensive care unit is generally advisable (1). Levels of ALA and porphobilinogen (PBG) can be monitored, and should decrease rapidly with Panhematin treatment. However, this biochemical response does not necessarily predict a clinical response, which will depend on how advanced the neuropathic manifestations were before treatment was started.
Supportive and Symptomatic Treatment: Harmful drugs should be stopped immediately and avoided during treatment. For example, metoclopramide should not be given. Information on harmful medications is found at the APF (www.apfdrugdatabase.com) and EPI websites (www.drugs-porphyria.org) as searchable databases that are updated periodically (1).
Intravenous glucose loading (at least 3 L of 10% glucose daily) should be used only for mild attacks (pain controlled with little or no narcotic analgesics, and without hyponatremia, motor weakness, etc.) or while waiting for Panhematin to become available. During treatment with hemin, large volumes of 10% glucose are not needed, which helps prevent dilutional hyponatremia. Nutrition support can be given using a central line and more concentrated solutions if needed during a prolonged attack.
Hyponatremia, hypomagnesemia and other electrolyte imbalances should be corrected and monitored. Safe drugs include narcotic analgesics for pain and phenothiazines for nausea, vomiting, anxiety or agitation. Beta-adrenergic blocking agents can be used to control the tachycardia and systemic arterial hypertension in patients without hypovolemia. Seizure precautions are recommended, especially in patients who develop hyponatremia. Treatment of seizures is difficult, since most commonly used anticonvulsants are harmful in acute porphyria. Gabapentin, benzodiazepines and vigabatrin are considered safe. Patients who experience seizures during an acute attack seldom require prolonged anticonvulsant therapy.
Patient education, genetic counseling, a medic alert bracelet or card, and consultation with a porphyria expert for additional advice and follow-up is recommended after recovery from an acute attack. The risk of hepatocellular carcinoma is increased in the acute porphyrias (6,7). Therefore, it is currently recommended that patients undergo screening by liver imaging for early detection at least yearly after age 50, especially if porphobilinogen (PBG) remains elevated.
The American Porphyria Foundation is a valuable source of additional advice. Patients should be encouraged to join and support this association.
Attacks recur in some patients, even if they avoid drugs and other factors that are known to cause exacerbations. Patients with frequent premenstrual attacks may benefit from treatment with a gonadotropin-releasing hormone analogue (8). A single infusion of Panhematin once or twice weekly may prevent noncyclic attacks in some patients, but experience is limited. Some patients treated repeatedly with hemin develop iron overload, which can be assessed by serum ferritin measurement, and treated by phlebotomies. Liver transplantation has been effective in a few patients with recurrent attacks that were not responsive to other interventions (2). Gene therapy may become available in the future.
Some patients develop chronic symptoms, including chronic pain syndrome and severe depression with an increase risk of suicide. These patients require careful supportive management, and their symptoms may improve over time.
*Please note that symptoms and treatment of HCP and VP are similar.
BIBLIOGRAPHY 1) Anderson KE, Bloomer, JR Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR and Desnick RJ. Recommendations for the Diagnosis and Treatment of the Acute Porphyrias. Ann Intern Med 2005; 142:439-450 2) Seth AK, Badminton MN, Mirza D, Russell S, Elias E, Liver transplantation for porphyria: Who, when, and how? Liver Transplantation. 2007;1219-1227 3) Deacon AC, Peters TJ, Identification of acute porphyria: evaluation of a commercial screening test for urinary porphobilinogen. Ann Clin Biochem. 1998;35:726-32 4) Blake D, McManus J, Cronin V, Ratnaike S. Fecal coproporphyrin isomers in hereditary coproporphyria. Clin Chem. 1992;38:96-100 5) Anderson KE, Bonkovsky HL, Bloomer JR and Shedlofsky SI. Reconstitution of he- matin for IV injection. Ann Intern Med 2006; 144:537-38 6) Andersson C, Bjersing L, Lithner F. The epidemiology of hepatocellular carcinoma in patients with acute intermittent porphyria. J Intern Med. 1996; 240:195-201 7) Andant C, Puy H, Faivre J, Deybach JC. Acute hepatic porphyrias and primary liver cancer [Letter]. N Eng J Med 1998; 338:1853-4 8) Anderson KE, Spitz IM, Bardin CW, Kappas A. A gonadotropin releasing hormone analogue prevents cyclical attacks of porphyria. Arch Intern Med. 1990; 150: 1469-74
Acute Intermittent Porphyrias Introduction PART 2
Tuesday - June 5, 2018 @ 16:27:57
INTRODUCTION
Porphyrias are inherited and/or acquired disorders of heme biosynthesis. A review article published in the March 2005 Annals of Internal Medicine (1) provides expert recommendations on the diagnosis and treatment of the acute porphyrias, which are the most life-threatening of the porphyrias. In addition, two valuable websites are available as resources for the ER physician:
Heme is synthesized in all tissues in eight enzymatic steps, and the end product is a component of many essential hemoproteins, such as hemoglobin, myoglobin and cytochromes, including the cytochrome P450 enzymes (CYPs) that are especially abundant in the liver. The first enzyme, ALA synthase (ALAS), controls the rate of heme synthesis in the liver. This enzyme is down-regulated by heme. The four acute hepatic porphyrias are due to different enzyme deficiencies (Table 1). The most common of these is acute intermittent porphyria (AIP), but the others can produce the same neurovisceral symptoms. The enzyme deficiency limits the capacity of the liver to increase heme synthesis. Therefore, when drugs, hormones or other factors that induce ALAS and CYPs are given, ALA and porphobilinogen (PBG) are overproduced and accumulate, and a neurovisceral attack may develop (2). Both ALA and porphobilinogen (PBG) increase during attacks of AIP, hereditary coproporphyria (HCP) and variegate porphyria (VP), although these increases may be less and more transient in HCP and VP. Increases in porphobilinogen (PBG) are generally greater than ALA. In ADP, which is extremely rare, only ALA is increased. Increases in urinary porphyrins also occur in these conditions, but are less specific because they also increase in many other medical conditions.
CLINICAL FEATURES
Gastrointestinal symptoms: Acute abdominal pain occurs in about 85-90% of attacks (1), and is neurologic in origin. The pain is usually severe, diffuse, unremitting for hours and poorly localized, but is sometimes colicky. Nausea, vomiting and constipation are common, but diarrhea is sometimes noted.
Neurologic symptoms: Pain in the extremities, back, chest or head is also common. Objective sensory loss may be found in up to 40% of cases (1). Peripheral motor neuropathy is an indication of a severe and potentially life-threatening attack. Neuropathy can progress to respiratory failure and bulbar paralysis in hours or days. Sudden death, presumably from cardiac arrhythmia may occur. Bladder paresis may cause dysuria and hesitancy (1).
The central nervous system is also affected. For example, insomnia is often an early symptom. Agitation, confusion, combativeness and other acute neuropsychiatric features are common and sometimes progress to coma. Convulsions may result from porphyria itself or from hyponatremia.
Physical findings: Tachycardia and systemic hypertension are very common. Fever is absent or mild. Signs of peritoneal irritation, such as tenderness are usually not prominent, but there is often ileus with distension and decreased bowel sounds. However, at times bowel sounds are normal or increased.
Motor weakness, which is usually symmetrical and begins proximally in the upper extremities, can be difficult to detect because common tests of strength, such as hand-grip, are not initially affected.
Clinical laboratory tests: Leukocytosis is usually absent or mild. Hyponatremia may reflect inappropriate antidiuretic hormone secretion. Gastrointestinal or renal sodium loss may contribute.
Sensitive and specific laboratory tests are available for diagnosis of acute porphyrias, especially during acute attacks, as described below.
Erythropoietic Protoporphyrias: Studies of the Natural History, Genotype-Phenotype Correlations, and Psychosocial Impact)
Erythropoietic Protoporphyrias: Studies of the Natural History, Genotype-Phenotype Correlations, and Psychosocial Impact)
How to participate:
In order to participate in a study, you must personally contact the APF
Edrin 1-866-APF-3635
Status: Recruiting
Background
The Erythropoietic Protoporphyrias include EPP and XLP. (Together they are the third most common porphyria and the most common in children.
The purpose of this study is to learn more about EPP and XLP and how to better treat people with them.
Researchers will observe participants over several years to learn about:
How often certain signs that indicate how bad the disease is occur as they are related to skin symptoms. For example, levels of protoporphyrins effects on the liver and other tissues affected by erythropoietic protoporphyrias.
What are the psychosocial effects of living with EPP/XLP.
About this Study
This is a longitudinal study (study taking place over 5 years) of about 150 individuals with EPP. Participants enrolled in this study should also be enrolled in the Longitudinal Study of the Porphyrias. Those participating in this study will be evaluated yearly. If possible, the first study visit should happen at one of the Porphyrias Consortium clinical sites (see the list of participating sites below).
Participation in this study will involve the following:
Accessing the information that you provided for the Longitudinal Study of the Porphyrias, including the results of the physical examinations, medical history, family history, questionnaires, and the results of laboratory tests;
Review of your medical records at each study visit; the study doctor may request medical records in between study visits to see how you are doing or if there is a medical concern that may be related to your porphyria or to the study;
Collection of samples, including:
blood (about 2-4 tablespoons)
urine (about ¼ cup) for testing and storage.
Completion of quality of life questionnaires
Testing on these samples may include measurement of certain products found in the blood and urine of people with porphyria and DNA analysis to find the mutation (or change) in the porphyria gene that is causing your porphyria. These samples will be collected at your first study visit, however, we may ask for additional samples later.
Have a diagnosis of erythropoietic protoporphyria (EPP) or X-Linked Protoporphyria (XLP)
You are not eligible to participate if you have:
You have elevations of porphyrins in urine, plasma or erythrocytes due to other diseases (i.e. secondary porphyrinuria or porphyrinemia), such as liver and bone marrow diseases.
A prior diagnosis of porphyria that cannot be documented by review of existing medical records or repeat biochemical or DNA testing.
Porphyria Post-The American Porphyria Foundation proudly announces our new office location!
Wednesday - May 30, 2018 @ 15:19:39
The APF is on the Move
The American Porphyria Foundation proudly announces our new office location!
Effective June 1, 2018, we will be located at:
American Porphyria Foundation 4915 St. Elmo Avenue, Suite 105 Bethesda, MD 20814 301-347-7166 866-APF-3635 Toll-Free www.porphyriafoundation.org
This move lands the APF near our nations capital and down the road from the NIH, the FDA, Capitol Hill and multiple national rare disease organizations. What it does not change is our relentless focus on impacting the lives of all individuals and families that live with Porphyria. We thank you for your tremendous support and look forward to serving you from our new home.
Participants are needed for an ongoing clinical trial for patients with PCT to study treatment with Harvoni, which is a prescription drug used to treat adults with chronic Hepatitis C. In this study all patients with PCT will be given a standard dose of Harvoni and will be monitored for two years.
If a participant is eligible, he/she will be asked to visit the study site once every month over the course of one year, and then once every 3 months for an additional year. At these visits the study doctors will check in with the participant and both blood and urine samples will be taken. ALL LABS and ALL MEDICATION are provided free of charge.
This is a wonderful opportunity for you to not only change your health, but to help change the health of others with PCT! Please contact Edrin Williams at the APF on 1-866-APF-3635 to connect with a research coordinator.
REMEMBERâ?¦RESEARCH IS THE KEY TO YOUR CURE!
Adrenal hormonal imbalance in acute intermittent porphyria patients: results of a case control study
Monday - May 21, 2018 @ 07:00:06
Adrenal hormonal imbalance in acute intermittent porphyria patients: results of a case control study
Acute Intermittent Porphyria (AIP) is a rare disease that results from a deficiency of hydroxymethylbilane synthase, the third enzyme of the heme biosynthetic pathway. AIP carriers are at risk of presenting acute life-threatening neurovisceral attacks. The disease induces overproduction of heme precursors in the liver and long-lasting deregulation of metabolic networks. The clinical history of AIP suggests a strong endocrine influence, being neurovisceral attacks more common in women than in men and very rare before puberty. To asses the hypothesis that steroidogenesis may be modified in AIP patients with biochemically active disease, we undertook a comprehensive analysis of the urinary steroid metabolome.
Methods
A casecontrol study was performed by collecting spot morning urine from 24 AIP patients and 24 healthy controls. Steroids in urine were quantified by liquid chromatography-tandem mass spectrometry. Parent steroids (17-hydroxyprogesterone; deoxycorticosterone; corticoesterone; 11-dehydrocorticosterone; cortisol and cortisone) and a large number of metabolites (N=55) were investigated. Correlations between the different steroids analyzed and biomarkers of porphyria biochemical status (urinary heme precursors) were also evaluated. The MannWhitney U test and Spearmans correlation with a two tailed test were used for statistical analyses.
Results
Forty-one steroids were found to be decreased in the urine of AIP patients (P<0.05), the decrease being more significant for steroids with a high degree of hydroxylation. Remarkably, 13 cortisol metabolites presented lower concentrations among AIP patients (P<0.01) whereas no significant differences were found in the main metabolites of cortisol precursors. Nine cortisol metabolites showed a significant negative correlation with heme precursors (p<0.05). Ratios between the main metabolites of 17-hydroxyprogesterone and cortisol showed positive correlations with heme-precursors (correlation coefficient>0.51, P<0.01).
Conclusions
Comprehensive study of the urinary steroid metabolome showed that AIP patients present an imbalance in adrenal steroidogenesis, affecting the biosynthesis of cortisol and resulting in decreased out-put of cortisol and metabolites. This may result from alterations of central origin and/or may originate in specific decreased enzymatic activity in the adrenal gland. An imbalance in steroidogenesis may be related to the maintenance of an active disease state among AIP patients.
Acute intermittent porphyria (AIP) is a dominant disorder that results from a partial deficiency of hydroxymethylbilane synthase (HMBS, EC 2.5.1.61) the third enzyme of the heme biosynthetic pathway[1]. Carriers of mutations within the HMBS gene are at risk of presenting acute life-threatening neurovisceral attacks[2]. The incidence of new symptomatic cases has been estimated to be 0.13 per million per year in most European countries[3].
The clinical presentation of AIP includes autonomous, central, motor and sensory symptoms. The patients may present abdominal pain, tachycardia, hypertension and hyponatremia. Neuropathy and muscle weakness can lead to tetraplegia, with respiratory and bulbar paralysis[4, 5]. Acute attacks are associated with the induction of 5-aminolevulinate synthase (ALAS-1), the first enzyme of the heme synthesis pathway, resulting in overproduction of heme precursors, aminolevulinic acid (ALA) and porphobilinogen (PBG) in the liver[6]. The exportation of ALA to tissues could be mainly responsible for the neurovisceral symptoms. However, the pathophysiology of the disease and the role of partial heme deficiency in tissues is not completely understood[7]. Acute attacks may be triggered by menstrual hormonal changes, fasting, stress and some therapeutic drugs. Intravenous heme administration is a well established highly effective therapy, albeit with transient effects[2]. Nevertheless, although heme administration may resolve acute crises, in most patients the urinary levels of PBG and ALA usually remain elevated for many years[8]. A few AIP patients develop recurrent acute attacks that may require repeated heme infusions or even liver transplantation for their cure[9].
The clinical history of AIP suggests a strong endocrine influence over disease expression, being neurovisceral attacks more common in women than in men and very rare before puberty or after menopause[7]. Moreover, in some series including few patients, the administration of gonadotropin releasing hormone analogues has been shown to reduce recurrent exacerbations associated with menstruation[10].
AIP patients with biochemically active disease present hepatic involvement with sustained ALAS-1 induction. In these patients, with long-lasting oveproduction of heme-precursors in the liver, we have previously reported low plasma levels of insulin-growth factor 1[11] and a decrease of 5α-reductase activity in the liver by the calculation of several urinary steroid metabolic ratios[12].
A pilot study by Larion et al.[13] studied circadian rhythms in AIP and found a decrease of plasma cortisol in patients with biochemically active disease. These findings suggested that in addition to hepatic involvement, AIP may be associated with hormonal disturbances originating in the adrenal gland. To test this hypothesis and in order to assess possible abnormalities in steroidogenesis among AIP patients, we undertook a comprehensive urinary target analysis of 70 steroid hormones and metabolites by state-of-the art liquid chromatographytandem mass spectrometry.
Methods
Patients
We studied 24 adult Caucasian Spanish patients with biochemically active AIP (22 women and 2 men, ranging in age from 22 to 54 years, Table 1). All these patients had initially presented an acute porphyria attack, had been diagnosed with AIP and regularly attended thereafter in the Porphyria Unit of the Hospital Clinic of Barcelona for clinical follow-up. AIP was assessed by biochemical and enzymatic analyses according to European Porphyria Initiative recommendations and external quality assessment schemes[14]. Genetic analysis of the HMBS synthase gene confirmed the AIP in all the cases. Some of the HMBS mutations found among these Spanish patients have been previously reported[15].
At the initiation of the study and prior to urine collection, the patients did not present symptoms of acute porphyria, but chronic complaints such as altered mood states, fatigue or pain in the back were frequently reported. Exceptionally, 3 patients who presented frequent recurrent attacks were on a prophylactic heme-arginate regime (Normosang; 3 mg/Kg; every 23 weeks). The urine of these patients was collected before the heme-arginate infusions. None of the patients included in the study were treated with luteinizing hormone-releasing hormone (LHRH) agonists.
Independently of the clinical status, all the patients presented increased long-term urinary excretion of the heme precursors PBG and ALA. The concentrations of the heme precursors in the urine samples used for steroid analysis are shown in Table 1.
None of the patients presented other diseases in addition to AIP, and all presented normal liver function. Although in some case-series AIP has been reported to be associated with chronic renal failure[16] which could eventually interfere with the objectives of the study, our series of Spanish AIP patients showed no evidence of an increased incidence of renal disease. All the cases included but one (with a borderline increase of creatinine concentrations in plasma) presented a normal glomerular filtration rate (GFR>60 mls/min, Table 1) estimated by the MDRD equation[17].
All AIP patients reported following the general dietetic and life-style recommendations for AIP carriers. Prior to urine collection all the patients were specifically interviewed and denied self-prescription or casual intake of porphyrinogenic drugs, synthetic hormones or other substances that could eventually interfere with steroid excretion and therefore bias the study results. Prior to steroid and porphyrin analyses urine integrity and normal pH values were checked in all the samples.
Twenty-four healthy volunteers (22 women and 2 men; age 2545 years) were recruited from the laboratory staff and included in the study as controls. They all presented normal renal function and were interviewed to discard the consumption of substances that could potentially interfere with steroid metabolism.
All patients and controls were informed of the purpose of the study and written consent was obtained. The research was conducted in accordance with the Declaration of Helsinki Principles and was approved by the Hospital Clinic Ethic Committee. Second morning urine from all patients and controls was obtained between 09.00 h-10.00 h in carefully controlled conditions within the hospital premises. Aliquots were immediately protected from light and frozen at -80°C until analyses.
PBG and ALA measurements
PBG and ALA were measured by ion-exchange chromatography using the ALA/PBG column test (Bio-Rad GmbH, Munich, Germany). Creatinine and liver enzymes were analyzed by standard methods using ADVIA 2400 equipment (Siemens Medical Solutions Diagnostics, Tarrytown, NY, USA). The concentration of PBG/ALA was normalized to creatinine (mmol/mol of creatinine).
Urinary steroids
Reference standards for steroid hormones, their metabolites and internal standards were obtained from Steraloids Inc. (Newport, USA), Sigma-Aldrich (St Louis, MO, USA), Merck (Darmstadt, Germany), Toronto Research Chemicals (Toronto, Canada), and NMI (Pymble, Australia) (for more information see reference[18]).
In summary, the target metabolomic analysis was based on the quantitation of urinary concentrations for 70 analytes including 17-hydroxyprogesterone (17OHP), 11-deoxycortisol (S), deoxycorticosterone (DOC), corticosterone (B), 11-dehydrocorticosterone (A), cortisol (F), cortisone (E) and testosterone (T) and a large number of their metabolites. These hormones represent key steps in steroidogenesis[19] (Figure 1). The structures of the urinary metabolites analyzed are summarized in Figure 2.
Figure 1
Biosynthesis of steroidal hormones and the enzymes involved.
Figure 2
Schematic representation of C21 steroidal hormone metabolism showing the analytes included in the study. Of the 70 analytes included in the analytical methods, 55 were detected in a substantial number of samples and therefore included in the evaluation of steroidogenesis in AIP patients. DHEA and T were also included in the study by the analysis of T, androsterone (from 5α-reduction+3α-reduction of DHEA and from 5α-reduction+3α-reduction+17-oxydation of T) and etiocholanolone (from 5β-reduction+3α-reduction of DHEA and from 5β-reduction+3α-reduction+17-oxydation of T) but have been omitted from the figure for clarity.
Quantification of urinary steroids
The quantification of urinary steroids was performed by a previously reported method[18]. Briefly, 0.5 mL of urine were added with an internal standard (ISTD) mixture, buffered at pH 7, and enzymatically hydrolysed with β-glucuronidase (60 minutes at 55°C). After hydrolysis, 2 mL of a saturated NaCl solution and 250 μL of a 25% (w/v) K2CO3 solution were added, and the mixture was extracted with 6 mL of ethylacetate. After evaporation of the organic layer, the extract containing glucuronoconjugated plus unconjugated analytes was reconstituted with 150 μL of water:acetonitrile (9:1, v/v). Unconjugated steroids were also determined by applying the same strategy but omitting the step of enzymatic hydrolysis.
Ten μL of the reconstituted extract were injected into the liquid chromatography-tandem mass spectrometry (LC-MS/MS) system consisting in a triple quadrupole (Xevo) mass spectrometer (Waters Associates, Milford, MA, USA) coupled to an Acquity UPLC system, (Waters Associates) for chromatographic separation. The LC separation was performed using an Acquity BEH C18 column (100 mm�2.1 mm i.d., 1.7 μm) (Waters Associates) at a flow rate of 300 μL min-1. Water and methanol both with formic acid (0.01% v/v) and ammonium formate (1 mM) were selected as mobile phase solvents. The detailed gradient and SRM method has been described elsewhere[18].
Quantification was performed after peak area integration of the analytes and the ISTD and comparison with a calibration curve. Results were normalized to creatinine levels (μg/g creatinine).
Statistical analysis
Among the metabolites determined by the study method, those detected in at least 50% of either the control or cases samples were included in the data analysis. The remaining analytes were discarded.
All the analytes were monitored in both unconjugated and total (conjugated+unconjugated) fractions. The unconjugated data was not considered for analysis among those analytes with a concentration in the unconjugated fraction representing less than 10% of the total amount, In cases in which the unconjugated concentration was greater than 90% of the total, the data obtained in the analysis of the total fraction was discarded from the analysis.
Data were analyzed using the SPSS software (v 18.0; IBM, Armonk, New York, NY, USA). The statistical analysis was used to reveal the differences between the two groups; cases and controls. Correlations between the different compounds analyzed, and the heme precursors (PBG and ALA) were also evaluated. Since the low number of data analyzed hampers the assumption of normality, the statistical analysis was conducted using non-parametric tests. A MannWhitney U test was used to compare the differences between the two groups and Spearmans correlation with a two-tailed test was used for the evaluation of the correlation among all the compounds analyzed. Statistical significance was set at pâ?¤0.05 or pâ?¤0.01.
A graphic representation including all the patients (Figure 3) was constructed by calculating the percentile of every sample for each analyte in relation to the combined population group (cases+controls). A gradual color scale plot was performed using green for the highest values (90% percentile or higher), yellow for intermediate values (50% percentile) and red for the lowest values (10% percentile or lower).
Figure 3
Graphical by-patient representation of the corrected urinary concentrations for the analytes studied. Green represents the 90% percentile, yellow the 50% percentile and red the 10% percentile. Lower concentrations (yellow-red tonalities) for cortisol and metabolites were found in AIP patients than in the control group (green-yellow tonalities). These differences were less significant in concentrations of cortisol precursors (17OHP, DOC and S) and their metabolites.
Results
Analytes included in the metabolomic study
Forty-eight analytes fulfilled the criteria for acceptance (see Statistical analysis section) and were therefore included in the metabolomic analysis. These compounds are summarized in Figure 2. Most were predominantly excreted (>90%) as conjugated with glucuronide whereas 5 metabolites (6βOH-B, 6βOH-F, 6βOH-E, 20α-DH17OHP and 20α-DHF) were detected mainly as unconjugated. Seven compounds (17OHP, F, E, 20βDHF, 20βDHE, 20αDHE and β,α-cortol) were detected (>10%) in both forms and therefore the concentrations obtained in the unconjugated and total (unconjugated+conjugated) fractions were considered separately for the evaluation. In summary, 55 analytes were considered for the metabolomic study of steroid profiling in AIP patients.
Case versus control groups
Case samples had heme precursor concentrations ranging from 3 to 64 nmol/mmol creatinine for PBG and from 1 to 40 nmol/mmol creatinine for ALA (normal values for PBG; <0.8; ALA<5).
The steroid concentrations are summarized in Table 2. The urinary concentrations of the steroid were within the normal range in the control samples[18, 20, 21, 22]. The concentrations found in the urine from AIP patients were generally lower for most of the analytes detected with significant differences (p<0.05) in 41 out of the 55 steroids evaluated. Remarkably, differences increased with the number of hydroxylation processes undergone by the hormone. Thus, no significant differences (p>0.05) were found in several metabolites of 17-hydroxyprogesterone, deoxycorticosterone and 11-deoxycortisol whereas 12 out of the 13 analytes related to cortisol (the most hydroxylated hormone) showed p values below 0.01 (the only exception was β, β-cortol with a p=0.021). Generally, the p values for the 5β reduced metabolites were higher than those for their 5α counterparts. Decreased excretion of 5α-steroids compared to their 5β-counterparts has been previously described[12, 23, 24, 25, 26].
Table 2
Results found in control and AIP patients for the hormones and metabolites determined
T + DHEA
17OHP
DOC
S
Control
AIP patients
Control
AIP patients
Control
AIP patients
Control
AIP patients
Parent
7.2
7.0
1.12
0.68
0.82
0.50
0.57
0.51
(5.5-16.4)
(5.3-10.3)
(1.09-1.69)
(0.65-1.16)**
(0.65-1.27)
(0.41-1.05)
(0.45-0.81)
(0.38-0.66)
u- Parent
n.d.
n.d.
0.77
0.58
n.d.
n.d.
n.d.
n.d.
(0.75-1.09)
(0.48-0.90)*
20β-red.
n.d.
n.d.
n.d.
n.d.
n.d.
n.d.
1.19
0.61
(1.06-1.49)
(0.56-1.0)**
u- 20α-red.
n.d.
n.d.
0.45
0.28
n.d.
n.d.
n.d.
n.d.
(0.39-0.57)
(0.20-0.42)**
5β-red. + 3α-red.
2924
1993
200
240
15.1
6.5
41.8
36.3
(25203636)
(16122642)**
(178382)
(218421)
(10.8-31.7)
(5.8-15.1)*
(40.8-71.4)
(33.8-50.2)
5α-red. + 3α-red.
3276
1262
n.d.
n.d.
2.4
0.1
n.d.
n.d.
(27563820)
(11351990)**
(2.7-7.6)
(0.22-1.27)**
5β-red. + 3α-red. + 20β-red.
n.d.
n.d.
983
1070
n.d.
n.d.
4.9
5.6
(9051346)
(8831493)
(4.7-11.7)
(3.9-8.4)
5β-red. + 3α-red. + 20α-red.
n.d.
n.d.
n.d.
n.d.
n.d.
n.d.
8.5
7.7
(7.0-12.5)
(6.6-10.5)
B
A
F
E
Control
AIP patients
Control
AIP patients
Control
AIP patients
Control
AIP patients
Parent
7.8
3.1
48.5
22.2
119
38.4
227
135
(7.0-14.6)
(2.7-6.9)**
(43.5-63.3)
(19.1-40.1)**
(91153)
(32.0-52.6)**
(211290)
(110173)**
u- Parent
n.d.
n.d.
n.d.
n.d.
50.7
10.3
106
43
(42.2-86.5)
(7.4-13.0)**
(99175)
(3969)**
u- 6β-hydroxyl.
3.7
0
n.d.
n.d.
291
87
17.2
10.7
(3.1-6.0)
(0.2-0.6)**
(265408)
(77122)**
(14.3-21.4)
(9.1-13.9)**
20β-red.
6.8
3.2
7.7
4.7
103
45
26.1
18.5
(5.2-7.7)
(2.5-4.6)**
(7.1-9.9)
(3.4-6.9)**
(88136)
(3764)**
(23.4-31.7)
(17.0-25.7)*
u- 20β-red.
n.d.
n.d.
n.d.
n.d.
62.3
24.3
13.2
7.2
(55.5-99.3)
(18.1-33.0)**
(10.0-17.9)
(6.0-10.4)**
20α-red.
5.4
3.3
17.0
8.8
n.d.
n.d.
64.0
39.3
(56.6-78.4)
(30.5-53.6)**
(5.1-10.3)
(2.8-5.8)*
(14.5-20.7)
(6.7-15.7)**
u- 20α-red.
n.d.
n.d.
n.d.
n.d.
230
36
38.1
17.8
(198363)
(3685)**
(34.2-52.6)
(14.3-27.0)**
5β-red.
n.d.
n.d.
n.d.
n.d.
6.9
4.3
9.4
13.0
(5.6-8.1)
(3.2-4.9)**
(6.3-16.4)
(8.2-19.0)
5β-red. + 3α-red.
190
87
29.6
16.9
2009
1115
3665
2876
(172271)
(69141)**
(24.7-40.5)
(14.0-31.6)*
(17512404)
(10401672)**
(30594325)
(25523743)
5α-red. + 3α-red.
308
89
8.1
2.8
967
350
56.6
31.8
(308470)
(89211)**
(7.5-12.2)
(2.4-5.0)**
(9121352)
(344884)**
(53.6-89.7)
(25.9-61.7)*
5β-red. + 3α-red. + 20β-red.
16.2
9.1
61.8
31.6
111
81
476
575
(9.9-25.0)
(7.2-20.0)
(50.7-91.7)
(32.7-94.8)
(94158)
(70115)*
(401597)
(512798)
5α-red. + 3α-red. + 20β-red.
n.d.
n.d.
n.d.
n.d.
35.4
14.0
n.d.
n.d.
(26.4-41.3)
(13.1-26.7)**
5β-red. + 3α-red. + 20α-red.
n.d.
n.d.
n.d.
n.d.
40.4
16.9
979
967
(29.8-50.9)
(14.8-31.8)**
(8301207)
(8271264)
5α-red. + 3α-red. + 20α-red.
n.d.
n.d.
n.d.
n.d.
59.8
22.3
n.d.
n.d.
(47.3-77.9)
(22.0-51.3)**
AIP patients exhibited a significant decrease for all cortisol metabolites whereas concentrations in the normal range were found for most the metabolites from cortisol precursors. Results are expressed as median (μg/g creatinine) and 95% confidence interval for mean (in brackets) *P<0.05, **P<0.01 vs. control, u- metabolite excreted unconjugated.
In order to check the individual status of every patient, Figure 3 provides a graphic representation of their steroid concentrations compared with the control group. For improving the clarity of the results, the patients were ordered by urinary PBG concentrations. In general, patients with relatively low heme precursor concentrations had urinary steroid concentrations closer to those of the control group, mainly showing yellow-orange tonalities in Figure 3. In contrast, AIP patients with higher concentrations of heme precursors had lower concentrations of steroids, showing predominantly orange-red tonalities in Figure 3. This decrease was more pronounced for cortisol, cortisone, corticosterone and 11-dehydrocorticosterone than for their 11-deoxycortisol, deoxycorticosterone and 17-hydorxyprogesterone precursors.
Correlation of steroids versus heme precursors
Based on the study by Christakoudi et al. in which steroid profiling was used for the study of 21-hydroxylase deficiency[27], a heat map was constructed showing the crossed correlations among the 55 urinary steroids, urinary PBG and ALA, found in AIP patients (Figure 4).This Figure, depicts the correlations found among all the analytes detected. Figure 5 provides the correlations found in AIP patients between heme precursors and the urinary steroids evaluated in greater detail. The correlation between cortisol and the remaining steroids is also included in Figure 5 as a model compound for steroid behaviour.
Figure 4
Crossed correlation found in AIP patients between the 55 urinary steroids, PBG and ALA. The expected positive correlation between hormones and their metabolites was obtained. Additionally, negative correlations between cortisol metabolites and heme precursors and positive correlations between 17-hydroxyprogesterone and heme precursors were found.
Figure 5
Correlations between heme precursors and the analytes selected. (a) correlation coefficients of PBG, ALA and F with all the analytes studied. Dotted and solid lines indicate a significance level at p=0.05 and p=0.01, respectively. The negative correlation between cortisol and heme precursors was of a similar magnitude to that of positive correlation obtained between cortisol and its direct precursor (corticosterone, B) or its direct product (cortisone, E). (b) correlation found between both heme precursors and the PT/5β-THF ratio (main metabolites of 17-hydroxy-progesterone and cortisol, respectively). The adrenal hormonal imbalance (ratio between the 17-hydroxy-progesterone precursor and the cortisol product) is correlated with the heme precursors detected in urine.
A positive correlation was found between cortisol and most of the urinary steroids, with this correlation being significant for most of the corticosterone, 11-dehydrocorticosterone, cortisol and cortisone metabolites evaluated (correlation coefficient between 0.4 and 0.9, p<0.05). The expected positive correlation between PBG and ALA was also observed (correlation coefficient 0.86, p<0.001).
Both heme precursors presented a negative correlation with most of the urinary steroids evaluated. The significance of this negative correlation increased with the number of hydroxylation steps performed in the biosynthesis of the hormone (see Figure 1), reaching a maximum for cortisol. Thus, 9 out of 14 cortisol metabolites showed a significant negative correlation with both PBG and ALA (correlation coefficient between 0.4 and 0.65, p<0.05). The lowest correlation between heme precursors and cortisol metabolites was found for 5α-reduced metabolites. In general, the lowest significance in the correlation was found for polyreduced metabolites.
A positive significant correlation was observed between PBG and pregnantriol; i.e. the main metabolite of 17-hydroxyprogesterone (correlation coefficient=0.482, p=0.02). This positive trend was also found for the second main 17-hydroxyprogesterone metabolite (17HP), although statistical significance was not achieved (correlation coefficient=0.334, p=0.1).
Since an opposite trend was found between the urinary metabolites of the first product of the metabolic cascade (17-hydroxyprogesterone) and those of the end product (cortisol), several ratios were established between 17-hydroxyprogesterone and the cortisol metabolites in order to check the global function of steroidogenesis. Since hepatic 5α-reduction is decreased in AIP patients[12, 23, 24, 25, 26] and no 5α-metabolites are included in 17-hydroxyprogesterone, the use of 5α-reduced metabolites was avoided and only 5β-reduced metabolites were considered for the formation of ratios. The ratio between the main metabolites of 17-hydroxyprogesterone and cortisol (PT/5β-THF) showed a positive correlation with both PBG and ALA (correlation coefficient>0.51, p<0.01, see Figure 5b). Similar results were obtained for the remaining ratios tested.
Discussion
The results presented in this study can be summarized into three main findings (i) urinary corticosteroid levels are decreased in AIP patients, (ii) cortisol and its metabolites are more decreased than its precursors in AIP, and (iii) the urinary concentration of cortisol metabolites is negatively correlated with the urinary concentration of heme precursors whereas an opposite trend may be observed for the main metabolites of 17-hydroxyprogesterone.
The study was performed with spot urine samples (second void of the morning) from AIP patient collected between 09.00 h and 10.00 h in a controlled hospital setting and not with at-home collected 24-hour urine. The possible weaknesses of a study based on spot-urine are minimized by the analysis of a large number of metabolites of each hormone. Since all cortisol metabolites were found to have a similar trend to a decrease in AIP patients (Table 2), our results seem to show a high degree of consistency, despite the use of spot morning urine.
The quantitation of a large number of hormones and metabolites also increased the significance of our findings. The measurement of a single metabolite (or a few metabolites) for each hormone would make the interpretation uncertain, since variations in urinary concentrations may be the consequence of either changes in the synthesis of a parent compound or changes in its metabolism (or a combination of both effects). In our study, every step of steroidogenesis was evaluated by different analytes, thereby increasing the reliability of the results as in the case of the decrease of cortisol which was supported by the decrease of 13 urinary species.
The overall decrease of corticosteroid excretion reported here is in agreement with the observations of Larion et al.[13] who found a decrease of cortisol in plasma in 3 AIP women with active disease. Moreover, we found a negative correlation between heme precursor concentrations in urine and most of the cortisol metabolites. Two tentative explanations can be hypothesized for our whole set of results: (i) AIP patients may present alterations in the hypothalamic pituitary adrenal (HPA) axis, and (ii) AIP patients may present deficiencies in the adrenal enzymes involved in the biotransformation of 17-hydroxyprogesterone to cortisol (Figure 1).
Neither of these hypotheses can be ruled out. However, if a decrease in pituitary-derived ACTH was the cause of the decrease in cortisol levels, a similar reduction would be expected for other adrenal corticosteroids. Our results showed normal values of 17-hydroxyprogesterone and 11-deoxycortisol metabolites suggesting that the adrenal activity was not reduced as a whole.
Light-sensitive extra-pituitary input from the suprachiasmatic nucleus regulated by the hippocampus induces a burst of cortisol secretion following morning awakening, the so-called cortisol awakening response (CAR)[28]. A possible dampening of CAR among AIP patients would also explain the decrease in cortisol observed in the morning urine. This effect would be more pronounced for those metabolites arising from rapid metabolism.
Acquired enzymatic deficiencies affecting the biosynthesis of cortisol could more likely explain the increased ratio between 17-hydroxyprogesterone metabolites (arising from the precursor) and cortisol metabolites (arising from the final product). This could be a consequence of heme deficiency associated with sustained up-regulation of ALAS-1 which is characteristic of active AIP. Heme deficiency could, in turn, partially decrease the activity of specific hemoproteins, notably P450 cytochromes, involved in steroid biosynthesis in the adrenal gland. Moreover, intracellular and mitochondrial energy deficiency, cofactor depletion or even direct ALA toxicity could also contribute to the changes observed in steroidogenesis.
It is unclear how the hormonal imbalance is associated with ALAS-1 induction and the sustained overproduction of heme precursors among AIP patients. It has been shown that steroids may induce ALAS-1 and porphyrin accumulation in liver cells[29]. Therefore, changes in adrenal metabolism could also be interpreted as physiological compensation mechanisms in situations of long-lasting ALAS-1 induction in the liver.
In conclusion, active AIP is associated with a hormonal imbalance of adrenal steroidogenesis; however, the effect of this imbalance on disease expression needs to be further evaluated.
Abbreviations
AIP:
Acute intermittent porphyria
HMBS:
Hydroxymethylbilane synthase
ALAS-1:
5-aminolevulinate synthase
ALA:
Aminolevulinic acid
PBG:
Porphobilinogen
17OHP:
17-hydroxyprogesterone
S:
11-deoxycortisol
DOC:
Deoxycorticosterone
B:
Corticosterone
A:
11-dehydrocorticosterone
F:
Cortisol
E:
Cortisone
T:
Testosterone
DHEA:
Dehydroepiandrosterone
DH:
Dihydrometabolite
TH:
Tetrahydrometabolite
6βOH:
6β-hydroxy-metabolite
u-:
Unconjugated metabolite
ISTD:
Internal standard
HPA:
Hypothalamic pituitary adrenal axis
ACTH:
Adrenocorticotopic hormone
CAR:
Cortisol awakening response.
Declarations
Acknowledgments
This work was supported by grants from Instituto de Salud Carlos III FEDER, (CP/10/00576) and the Spanish Fondo de Investigación Sanitaria (PI11/00767) to Jordi To-Figueras. Technical support of Nuria Renau is acknowledged.
Anderson KE, Sassa SS, Bishop DF, Desnick RJ: Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. The metabolic basis of inherited disease. 8th edition. Edited by: Scriver CR, Beaudet AL, Sly WS, Valle D. New York: McGraw-Hill; 2001.Google Scholar
Elder G, Harper P, Badminton M, Sandberg S, Deybach JC: The incidence of inherited porphyrias in Europe. J Inherit Metab Dis. 2013, 36 (5): 849-857. 10.1007/s10545-012-9544-4.View ArticlePubMedGoogle Scholar
Kauppinen R, Mustajoki P: Prognosis of acute porphyria: occurrence of acute attacks, precipitating factors, and associated diseases. Medicine (Baltimore). 1992, 71: 1-13.View ArticleGoogle Scholar
Hift RJ, Meissner PN: An analysis of 112 acute porphyric attacks in Cape Town, South Africa: evidence that acute intermittent porphyria and variegate porphyria. Medicine (Baltimore). 2005, 84: 48-60. 10.1097/01.md.0000152454.56435.f3.View ArticleGoogle Scholar
Thunell S: Outside the box: genomic approach to acute porphyria. Physiol Res. 2006, 55: 43-66.Google Scholar
Marsden JT, Rees D: Urinary excretion of porphyrins, porphobilinogen and δ-aminolaevulinic acid following an attack of acute intermittent porphyria. J Clin Pathol. 2014, 67: 60-65. 10.1136/jclinpath-2012-201367.View ArticlePubMedGoogle Scholar
Soonawalla ZF, Orug T, Badminton MN, Elder GH, Rhodes JM, Bramhall SR, Elias E: Liver transplantation as a cure for acute intermittent porphyria. Lancet. 2004, 363: 705-706. 10.1016/S0140-6736(04)15646-8.View ArticlePubMedGoogle Scholar
Anderson KE, Spitz IM, Sassa S, Bardin CW, Kappas A: Prevention of cyclical attacks of acute intermittent porphyria with a long-acting agonist of luteinizing hormone releasing hormone. N Engl J Med. 1984, 311: 643-645. 10.1056/NEJM198409063111006.View ArticlePubMedGoogle Scholar
Deybach JC, Casamitjana R, Puy H, Herrero C: Role of two nutritional hepatic markers (insulin-like growth factor 1 and transthyretin) in the clinical assessment and follow-up of acute intermittent porphyria patients. J Intern Med. 2009, 266: 277-285. 10.1111/j.1365-2796.2009.02118.x.View ArticlePubMedGoogle Scholar
Casals G, Marcos J, Pozo OJ, Aguilera P, Herrero C, To-Figueras J: Gas chromatographymass spectrometry profiling of steroids in urine of patients with acute intermittent porphyria. Clin Biochem. 2013, 46 (9): 819-824. 10.1016/j.clinbiochem.2013.03.001.View ArticlePubMedGoogle Scholar
Larion S, Caballes FR, Hwang SI, Lee JG, Rossman WE, Parsons J, Steuerwald N, Li T, Maddukuri V, Groseclose G, Finkielstein CV, Bonkovsky HL: Circadian rhythms in acute intermittent porphyriaa pilot study. Eur J Clin Invest. 2013, 43 (7): 727-739. 10.1111/eci.12102.View ArticlePubMedPubMed CentralGoogle Scholar
Aarsand AK, Villanger JH, Støle E, Deybach JC, Marsden J, To-Figueras J, Badminton M, Elder GH, Sandberg S: European specialist porphyria laboratories: diagnostic strategies, analytical quality, clinical interpretation, and reporting as assessed by an external quality assurance program. Clin Chem. 2011, 57 (11): 1514-1523. 10.1373/clinchem.2011.170357.View ArticlePubMedGoogle Scholar
To-Figueras J, Badenas C, Carrera C, Muñoz C, Mila M, Lecha M, Herrero C: Genetic and biochemical characterization of 16 acute intermittent porphyria cases with a high prevalence of the R173W mutation. J Inherit Metab Dis. 2006, 29 (4): 580-585. 10.1007/s10545-006-0344-6.View ArticlePubMedGoogle Scholar
Marsden JT, Chowdhury P, Wang J, Deacon A, Dutt N, Peters TJ, Macdougall IC: Acute intermittent porphyria and chronic renal failure. Clin Nephrol. 2008, 69 (5): 339-346. 10.5414/CNP69339.View ArticlePubMedGoogle Scholar
Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D: A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Ann Intern Med. 1999, 130 (6): 461-10.7326/0003-4819-130-6-199903160-00002.View ArticlePubMedGoogle Scholar
Marcos J, Renau N, Casals G, Segura J, Ventura R, Pozo OJ: Investigation of endogenous corticosteroids profiles in human urine based on liquid chromatography tandem mass spectrometry. Anal Chim Acta. 2014, 812: 92-104.View ArticlePubMedGoogle Scholar
Miller WL, Auchus RJ: The molecular biology, biochemistry and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011, 32 (1): 81-151. 10.1210/er.2010-0013.View ArticlePubMedPubMed CentralGoogle Scholar
Weykamp CW, Penders TJ, Schmidt NA, Borburgh AJ, van de Calseyde JF, Wolthers BJ: Steroid profile for urine: reference values. Clin Chem. 1989, 35: 2281-2284.PubMedGoogle Scholar
Fischer DA: The Quest Diagnostic Manual, endocrinology. Test selection and interpretation. Quest Diagnostics Incorporated. 2007, 105.Google Scholar
Shackleton CHL, Marcos P: GC-MS steroid Profiling: Diagnosis of disorders affecting steroid synthesis and metabolism. The Encyclopedia of Mass Spectrometry. Edited by: Gross M, Caprioli R. 2006, Amsterdam: Elsevier, 789-813.Google Scholar
Kappas A, Bradlow HL, Gillette PN, Gallagher TF: Studies in porphyria. I. A defect in the reductive transformation of natural steroid hormones in the hereditary liver disease, acute intermittent porphyria. J Exp Med. 1972, 136: 1043-1053. 10.1084/jem.136.5.1043.View ArticlePubMedPubMed CentralGoogle Scholar
Bradlow HL, Gillette PN, Gallagher TF, Kappas A: Studies in porphyria. II. Evidence for a deficiency of steroid delta-4-5-alpha-reductase activity in acute intermittent porphyria. J Exp Med. 1973, 138: 754-763. 10.1084/jem.138.4.754.View ArticlePubMedPubMed CentralGoogle Scholar
Christakoudi S, Deacon AC, Peters TJ, Taylor NF: Urinary steroid hormone metabolites in patients with porphyria. Endocrine Abstracts. 2003, 5: 241.Google Scholar
Innala E, Bäckström T, Poromaa IS, Andersson C, Bixo M: Women with acute intermittent porphyria have a defect in 5α-steroid production during the menstrual cycle. Acta Obstet Gynecol Scand. 2012, 91: 1445-1452. 10.1111/j.1600-0412.2012.01536.x.View ArticlePubMedGoogle Scholar
Christakoudi S, Cowan DA, Christakoudis G, Taylor NF: 21-hydroxylase deficiency in the neonate-trends in steroid anabolism and catabolism during the first weeks of life. J Steroid Biochem Mol Biol. 2013, 138: 334-347.View ArticlePubMedGoogle Scholar
Clow A, Hucklebridge F, Stalder T, Evans P, Thorn L: The cortisol awakening response: more than a measure of HPA axis function. Neurosci Biobehav Rev. 2010, 35: 97-103. 10.1016/j.neubiorev.2009.12.011.View ArticlePubMedGoogle Scholar
Stephens JK, Fisher PV, Marks GS: Porphyrin induction: equivalent effects of 5alphaH and 5betaH steroids in chick embryo liver cells. Science. 1977, 197 (4304): 659-660. 10.1126/science.877578.View ArticlePubMedGoogle Scholar
Athi Koti AIP Experience
Thursday - May 17, 2018 @ 07:00:08
Type of Porphyria:
Acute Intermittent Porphyria (AIP)
Sive Ngalo of Ekasi Hub interviewed Athi Koti.
Below is my story
Athi Koti is a 30 year old physically challenged man, originally from Eastern Cape in Bhisho. He embarked on his journey by pursuing the IT (Information Technology) diploma at Walter Sisulu University in Butterworth in 2008 and completed it in 2011. On the 9th of May 2012 it was his graduation day, a day of celebration for his achievement towards finishing his studies in record time, but that day turned to be a date of sadness, pain, grief, and hopelessness. He woke up early that day due to excitement but then he realized that something was not right as he was about to get up and brush his teeth, trying to get off the bed he felt cramps on his legs and in that struggle he fell as his legs became numb. He then called upon his brother Sandiso and he came rushing to the room and shockingly asked him what was wrong and he carried him back to his bed.
I asked Sandiso (My little brother) to phone my mother and tell her that my lower limbs (legs) are weak and I cannot walk, and he did so. My mother then called my uncle who then took me to Bhisho Hospital. I was admitted immediately, that time I felt all sorts of emotions. After 3 days in hospital my upper limbs (arms) also got affected and stop moving then I became bedridden. On the fifth day I was transferred to another private Hospital for spinal cord scan because I could not sit, all my body joints were not moving. At the private Hospital I went to for my spinal cord scan, the results came back negative which meant that my spinal cord was normal.
I was then transferred to Cecilia Makiwane situated in Mdantsane. After a week I decided to go home since I was just sleeping in bed without being taken to Physiotherapy sessions. I was praying, crying, hopeless, and I ended up asking my mother to ask the mother of my son (who was 4 months old that time) to bring him all the way from Pietermaritzburg, because it seemed like my days were numbered and I would suddenly pass on the following day.
My son came, and I was stressed because when he was crying I couldn't hold him, touch him and feel or bond with him because my whole body was numb. They stayed only for March 2012 and again I was transferred from Eastern Cape to Western Cape (Groote Schuur Hospital). I arrived there on the 26 of April 2012 then doctors did a lumber puncher to diagnose my illness and they discovered that I had an Acute Intermittent Porphyria Neuropathy (Nervous system is not functioning well) but they do not know the cause of it and they said it's genetic but both my parents tested negative and there was no family history of this kind of illness. The prognosis was that I was going to be in a wheel chair for 3 years, but since there is nothing impossible with God, I have managed to walk within 3 months before they transferred me to Western Cape Rehabilitation Centre in Lentegeur (Mitchell's Plain). And that is when I discovered that I was permanently disabled.
It's true that when days are dark friends are few. It was Akhona Dyantyi (A friend of mine) who stood by my side throughout the entire ordeal, making an effort to comfort and give me hope. I was also going to church to praise the Lord for proving the doctors wrong. Despite it all, God blessed me with a job and two sons, from a wheelchair and now walking with crutches. Now I believe that "In life you must not live in a situation but you must live above the situation" by Pastor Sithole.
My work journey has not been a pleasant one but I was fortunate enough to get into Transnet Port Terminals when they offered a learnership to disabled people, as that enabled me to provide for my children. Unfortunately I could not study further due to my disability. After my learnership contract had expired I was then again fortunate to have landed a permanent position, in Cape Town as a Junior Analyst in the Management Information Systems department (MIS).
And the winner is..
Friday - May 11, 2018 @ 07:00:02
May 16, 2018
PORPHYRIA POST
2018 President's Award
Each year, the American Porphyria Foundation selects a recipient who has gone above and beyond in support of our foundation and the porphyria patient community. We are proud to present the following individuals with the President's Award 2018.
(L to R) Brooklyn, Jared, Krista and Hailey
Congratulations to.
Nicole Castellano
Nicole is a champion for people who live with porphyria. She is exemplary at managing a difficult disease with a can-do attitude not only for herself, but by encouraging others. Nicole trained and ran the Chicago Marathon, never an easy task, let alone while managing AIP. She used the experience to raise funds for the APF and every step of her training was filled with honesty and awareness (and videos!) about the burden of living with AIP. Thank you, Nicole, for all you have done to educate others and raise awareness!
Jared Ulmer
You may know him as Porphyria J! Jared has used his artistic talent and lively sense of humor to create the Porphyria J video series (vlog), focused on creating community and awareness around Porphyria. Available on the APF YouTube channel, each video focuses on a different area of living with EPP What is Porphyria? A Day in My EPP Live! Not Alone. Travelling with EPPand many more. Jared also ran the Super Spartan Race of course, he archived that experience! Thank you, Jared - we look forward to where your creativity takes you next. Until then, Seek Shade and Stay Happy!
The word erythropoietic means associated with red blood cells (erythro-) and their formation (-poietic). The porphyrias are a group of uncommon diseases caused by something going wrong with the production of chemicals known as porphyrins. These chemicals are the building blocks of haem, which, when combined with a protein (globin), forms haemoglobin, the material in red blood cells that carries oxygen round the body. In the case of EPP, there is a build up of one of these porphyrins (protoporphyrin) in the blood, especially in the red blood cells. This leads to a sensitivity to sunlight.
What causes EPP?
An enzyme is a protein that helps to convert one chemical substance into another. In EPP, there is a shortage of one particular enzyme (ferrochelatase), which normally helps to convert protoporphyrin into haem by adding iron to it. As a result of this enzyme deficiency, protoporphyrin levels build up in the blood. As blood passes through the skin, the protoporphyrin absorbs the energy from sunlight and this sets off a chemical reaction that can slightly damage surrounding tissues. The nerve endings in the skin interpret this as itching or burning pain, and if the blood vessels are affected, they can leak fluid, causing swelling.
The light that protoporphyrin absorbs is different from that which causes ordinary sunburn. Usually sunburn is caused by the shorter wavelengths of ultraviolet light (UVB), but in EPP the skin is more sensitive to longer ultraviolet wavelengths (UVA) and to visible light.
Is EPP hereditary?
Yes, but there is not always a family history of the condition. Everyone has two genes for ferrochelatase in each cell in their body (one coming from their mother and one from their father). In most families, EPP occurs when an affected individual inherits a gene for a severely underactive ferrochelatase enzyme from one parent, and a less severely affected gene from the other parent. The less severely affected gene is quite common, being present in about 10% of the general population, but it never causes EPP by itself. The genetics is quite complex and advice from your local genetics service may be useful.
What are the symptoms of EPP?
Typically EPP starts with abnormal sensitivity to sunlight. Exposure to sunlight causes tingling, itching or burning, which may be associated with redness and swelling. These symptoms usually occur within a few minutes of skin exposure to sunlight, and often they take hours or days to resolve. During this time the skin may feel more sensitive than usual to extremes of temperature. The light producing these changes need not be direct light reflected off water and sand, or passing through window glass, including car windscreens, can also cause the symptoms.
EPP usually starts in childhood, and affects males and females equally. Infants may cry or scream after being taken out into the sunlight; and older children may complain of burning and try to wave their hands in the air, or put them into cold water to try to relieve the pain. A very small number of people who have had with EPP for many years may develop liver damage. Fortunately this is rare.
What does EPP look like?
Despite severe discomfort, there may be nothing abnormal to see on the skin. Sometimes there can be swelling of the skin, initially like a nettle rash. With time, some people develop thickening of the skin over their knuckles, and small scars on sun-exposed skin such as that on the cheeks, nose, and backs of the hands. However these skin changes show wide variation between different individuals.
How is EPP diagnosed?
The diagnosis is usually suspected from the story, and can be confirmed by a blood test. This measures the amount of protoporphyrin in the blood (serum protoporphyrin) and in the red blood cells (erythrocyte free protoporphyrin). Some doctors will also ask for a stool sample to measure the level of protoporphyrin in the faeces. No urine tests are relevant to this condition except to exclude other types of porphyria.
Although it is unlikely that you will develop liver problems as a complication of EPP, your doctor may monitor the way your liver is working by yearly blood tests. If there is any evidence of a deterioration in liver function, there are certain interventions that may help to halt or reverse this.
As EPP affects the production of haemoglobin, it is not uncommon for people with EPP to be slightly anaemic. Your doctor will probably also measure your blood count to make sure that you are not becoming too anaemic.
Can EPP be cured?
At present there is no cure for EPP.
For information on available treatments please go tothis pageon the website of the British Association of Dermatologists
Learn about Kimberly McIntyre and how EPP affects her daily!
Friday - May 4, 2018 @ 07:30:15
Name: Kimberly McIntyre Age: 34
â?¢ How old were you when you were diagnosed?
27
How long did it take you before finding out that you had Porphyria?
22 years
Was testing easy or prove to be difficult? If so in what ways?
It was easy once we knew what to test for
â?¢ Do you remember your first flare/reaction?
No, I was 3 years old
â?¢ What did it feel like to you?
It feels like Im burning from the inside out
â?¢ What things help you feel better? (Cool water, ice, shade, bath, clothes)
If Im able to, a cool fan usually Im just in too much PAIN!
â?¢ How long does it take before you start to feel better?
The first three days are really bad, and it takes a full week to be pain free
â?¢ What kind of clothing/trends do you wear when you go outside or in bad lighting? Always needing long sleeves shirts, but the heat triggers me as well.
â?¢ Do you enjoy being outside?
I love it but not when its sunny.
â?¢ What ways are you able to adapt to do certain activities outside?
I typically stay in the shade even then its risky.
â?¢ What fun things are you able to do inside your home while the sun is out?
I enjoy watching movies with my kids.
â?¢ Do you vacation or travel? Is this easy for you?What difficulty do you face?
Having to cover up and I am limited to what I can do. Parks are hard because of the lines and the rides are not in the shade and sometimes the wind will trigger me.
â?¢ Have you met any other Adults with EPP before?
My sister has EPP to.
â?¢ What things do you have to do to protect your skin?
Staying completely out of the sun.
â?¢ Is it hard for you to tell your friends, family what EPP is?
was when I was a child I was embarrassed now I dont care. Most people dont believeme.My daughter has it and I dont want her to have the feelings I had about myself.
â?¢ What do you tell them?
I tell them I am a vampire (NOT REALLY) to try to lighten the mood. Then tell them I am allergic to the sun and it hurts more than you can imagine to be in it
â?¢ As an Adult do you ever feel left out, with activities?
It hurts when my son wants my daughter and myself to play with him outside on a sunny day and were not able to.
How does this make you Feel?
Honestly, I feel horrible.
Do you ever face depression or sadness?
I used to there was one-point I had a bad reaction and I just wanted to give up.I didnt want to live this life, but now that I am an adult I realize I am stronger then this and I need to be for my kids.
Do you work?
Yes
What type of work do you do?
Wearhouse
Is this a challenge with Sun or Lights?
No, not with sun or lights but the pain and tiredness.
Has your work had to make changes for you to continue working without an EPP reaction?
No
â?¢ What advice can you share to help other Adults that have EPP?
Talk to others with it everyone is different and every time its youre trigger time it may be different. We understand, and we are here for each other.
â?¢ Would you benefit from a new treatment?
It would be life changing
Thank you, Kim, for sharing an adult perspective on EPP.We hope that a treatment will be approved soon for everyone with EPP to benefit.
Rare Artist Contest
Thursday - May 3, 2018 @ 06:30:05
News Feed
The EveryLife Foundation for Rare Diseases is excited to announce that the tenth annual Rare Artist contest is now open for submissions. This contest, which celebrates the unique talents of the rare disease community, is open to rare disease patients, family members, friends, caregivers and medical professionals. Changes from past contests are as follows:
The EveryLife Foundation for Rare Diseases is excited to announce that the tenth annual Rare Artist contest is now open for submissions. This contest, which celebrates the unique talents of the rare disease community, is open to rare disease patients, family members, friends, caregivers and medical professionals.
Changes from past contests are as follows:
â?¢ Entrants may now submit TWO entries in one of the four categories. Entrants may only win one award in their chosen category. â?¢ We have upgraded our system this year to ensure votes are tabulated accurately. Each vote is now logged to the users account, so we can track voting and ensure all numbers are accurate. The contest will accept entries through October 19th, and visitors to the Facebook gallery can vote for their favorites once every ten days through October 26th. Two awards will be given in each category: one to the entry receiving the most votes on Facebook, and the other selected by a panel of rare disease community leaders. Award recipients in each of the four categories will receive the following prizes: Children (4-11): $100 gift card Teens (12-18): $250 gift card Adults (19+): $500 gift card Adults (19+) - Photography/Digital Art: $500 gift card
Award recipients will have their work displayed during Rare Disease Week on Capitol Hill at the Rare Artist reception in Washington, DC on February 27th, 2019. Artists with works chosen for this reception will be eligible for travel stipends to provide the opportunity to see their work displayed on Capitol Hill. For additional information including the governing rules, please visit the contest rules page. If you have questions about the contest, please email the contest curator, Grant Kerber,
Welcome MAY News Reminders
Saturday - April 28, 2018 @ 12:40:02
Welcome members to the American Porphyria Foundation Purple Light Blog
Watch for New Members Stories & Interviews
If you would like to share your story please email me Amy@porphyriafoundation.org
Watch for Research Search Opportunities
Do you need help finding a Doctor?
Need a Comprehensive Dr. Kit?
Do you need a Patient Packet?
Learn whats in the Patient Packet
AIP, HCP, VP Access to Care Toolkit What & Where to find it?
#NPAW 2018 Meet Ben Edwards A Youths Interview With EPP
Friday - April 27, 2018 @ 12:30:03
#PAW2018
·
Ben Edwards Age: 10
How old were you when you were diagnosed? I was two years old.
Do you remember your first flare/reaction? I was at the beach building a sand castle near an umbrella, I was getting really agitated.I then started to feel horrible and then bad, I looked down at my hands they were swelling up and turning to a red & purple color-like a rash.
What did it feel like to you?I felt like everything I was touching was burning my skin.
What things help you feel better? (cool water, ice, shade, bath, clothes) Having a wet substance/cloth around my hands or anywhere my body hurts.I have even used wet socks before and now I use the Frog Toggs those stay cold longer.Putting it under water and blowing on it when I dont have water near me.Also using a fan.
How long does it take before you start to feel better?A few days, but my first and worst reaction took a week for it to go away.
What kind of clothing/trends do you wear when you go outside or in bad lighting? I always wear long sleeves no matter the temperature outside. I also wear pants. If its hot outside, I try to wear shirts with wicking fabric that keeps me cool. I use a hat with a flap on the back to cover my neck, even though I dont enjoy wearing it. In the snow I use a face mask. I wear gloves to cover my hands from the sun, especially when I stay outside for a long time.
What is your most favorite sport to play or watch? Do you play it inside or outside? During the day or at night? I love to participate on the swim team/swimming. I get to swim indoors and at night, but swim practice is early enough in the morning that it doesnt bother me to much.
What ways are you able to adapt to do certain activities outside?I try to stay in the shade& wear the right clothing. For instance, when I go to Glenwood Springs School their pool is open until 10pm so I can swim at night.
What fun things are you able to do inside your home while the sun is out?I like to build with my Legos, watch movies, play video games, read, write and draw.
What has been the best vacation ever?I loved Legoland. But my EPP was bad there, the sun hurt my hands, so I was not able to enjoy the Park as much as I wanted to.
Have you met any other kids with EPP before?No
Do you go to School/Homeschool?Elementary School
Do you have to go by car, bus or walk? Bus
What things do you have to do to protect your skin (In school) or (taking the car or bus)?For school and taking the bus, I wear long sleeves and long pants. My mom insists on me wearing a hat and gloves, at recess in the spring and fall, I dont want to but I know its for my protection. My hands always seem to end up hurting the most when its bright outside. In the winter its not too bad for me.
Is it hard for you to tell your friends, teachers or family what EPP is?No, not hard, they always forget because I have a mild case.
What do you tell them?I tell them I have a condition where if Im out in the sun to long my hands will start itching and swelling up. It gets bad and swollen- I just dont like that. When I was younger I used to tell people, I was allergic to the sun because they understood that a bit better. My mom sends information from the APF to my teachers & share Jared Ulmers videos about EPP.
What things do you do if you feel sad or left out?I sit in the shade with a friend, or I go play video games.
What do you want to be/do when you grow up?I want to be a video game programmer
What advice can you share to help other kids that have EPP?Stay in the SHADE! When my Dad is walking the dog in the park, I will jump from one shady spot to another.
Parents- How do your children deal with their EPP? (flare/pain)?As Ben gets older, he doesnt tell us how often hes in pain, so hes deals with it on his own. If hes really hurting he asks for a wet cold cloth or fan.
Would you benefit from a new treatment? If so how would it help you?Yes, I would if it doesnt change my appearance. I dont want to be tan. It would also help me to be out in the sun more, especially at swim meets & swim team. Id love to be able to go outside and not feel pain. I could go to places that I enjoy like LEGOLAND, always being able to do whatever I wanted to outside.
Thank you Ben for taking the time and sharing your examples of how you deal with EPP! Enjoy your swimming and swim team! I know youll be a great Gaming Programmer.
#PAW 2018 Medical Moment~ VP Variegate Porphyria
Friday - April 27, 2018 @ 07:00:05
#PAW2018
Variegate porphyria
Other Names:
Porphyria variegate; VP; Porphyria, South African type; See More
Variegate porphyria is a form of hepatic porphyria most common in the white South African population. This autosomal dominant disorder may produce acute attacks (as in acute intermittent porphyria) as well as skin photosensitivity.[1][2] The condition is caused by mutations in the PPOXgene which lead to deficiency of the enzyme protoporphyrinogen oxidase.[3] Acute attacks are managed and may be prevented as in acute intermittent porphyria.[1]
Episodes of acute variegate porphyria can cause abdominal pain, vomiting, constipation, and diarrhea. During an episode, a person may also experience muscle weakness, seizures, fever, and mental changes such as anxiety and hallucinations. Individuals with variegate porphyria may also develop cutaneous symptoms, including skin photosensitivity.[3]
Last updated: 7/19/2010
This table lists symptoms that people with this disease may have. For most diseases, symptoms will vary from person to person. People with the same disease may not have all the symptoms listed. This information comes from a database called the Human Phenotype Ontology (HPO) . The HPO collects information on symptoms that have been described in medical resources. The HPO is updated regularly. Use the HPO ID to access more in-depth information about a symptom.
Variegate porphyria is caused by mutations in the PPOXgene.[3] Mutations in the PPOX gene reduce the activity of protoporphyrinogen oxidase, allowing compounds called porphyrin precursors to build up in the body. These compounds are formed during the normal process of heme production, but reduced activity of protoporphyrinogen oxidase allows them to accumulate to toxic levels. More than 130 mutations in the PPOX gene have been identified in people with variegate porphyria. A particular PPOX gene mutation is found in about 95 percent of South African families with the disorder.[4]
Nongenetic factors such as certain drugs, alcohol, dieting, as well as other genetic factors that have not been identified, also contribute to the characteristic features of variegate porphyria.[4]
Variegate porphyria is inherited in an autosomal dominant manner, which means one copy of the gene in each cell is mutated. This single mutation reduces the activity of an enzyme needed for heme production, which increases the risk of developing signs and symptoms of porphyria.[1][3]
The diagnosis of variegate porphyria is made by finding excess coproporphyrin in urine and both coproporphyrin and protoporphyrin in feces.[1][2] The most sensitive screening test is probably a plasma porphyrin assay.[1]
Last updated: 7/19/2010
Testing Resources
The Genetic Testing Registry (GTR) provides information about the genetic tests for this condition. The intended audience for the GTR is health care providers and researchers. Patients and consumers with specific questions about a genetic test should contact a health care provider or a genetics professional.
Acute attacks are managed and may be prevented as in acute intermittent porphyria.[1][2] Hospitalization is often necessary for acute attacks. Medications for pain, nausea and vomiting, and close observation are generally required. A high intake of glucose or other carbohydrates can help suppress disease activity and can be given by vein or by mouth. Intravenous heme therapy is more potent in suppressing disease activity. It can be started after a trial of glucose therapy. However, the response to heme therapy is best if started early in an attack. Heme must be administered by vein. Panhematin is the only commercially available heme therapy for treatment and prevention of acute porphyric attacks in the United States. Heme arginate, which is marketed in some other countries, is another preparation of heme for intravenous administration.[5]
Last updated: 7/19/2010
Management Guidelines
Orphanet Emergency Guidelines is an article which is expert-authored and peer-reviewed that is intended to guide health care professionals in emergency situations involving this condition.
The American Porphyria Foundation offers a document that includes information about porphyria, types, testing, and treatment with Panhematin. Click the "document" link above to view these guidelines.
If you need medical advice, you can look for doctors or other healthcare professionals who have experience with this disease. You may find these specialists through advocacy organizations, clinical trials, or articles published in medical journals. You may also want to contact a university or tertiary medical center in your area, because these centers tend to see more complex cases and have the latest technology and treatments.
If you cant find a specialist in your local area, try contacting national or international specialists. They may be able to refer you to someone they know through conferences or research efforts. Some specialists may be willing to consult with you or your local doctors over the phone or by email if you can't travel to them for care.
You can find more tips in our guide, How to Find a Disease Specialist. We also encourage you to explore the rest of this page to find resources that can help you find specialists.
Research helps us better understand diseases and can lead to advances in diagnosis and treatment. This section provides resources to help you learn about medical research and ways to get involved.
Patient Registry
The Porphyrias Consortium is a team of doctors, nurses, research coordinators, and research labs throughout the U.S., working together to improve the lives of people with this condition through research. The Porphyrias Consortium has a registry for patients who wish to be contacted about clinical research opportunities.
Support and advocacy groups can help you connect with other patients and families, and they can provide valuable services. Many develop patient-centered information and are the driving force behind research for better treatments and possible cures. They can direct you to research, resources, and services. Many organizations also have experts who serve as medical advisors or provide lists of doctors/clinics. Visit the groups website or contact them to learn about the services they offer. Inclusion on this list is not an endorsement by GARD.
RareConnect has an online community for patients and families with this condition so they can connect with others and share their experiences living with a rare disease. The project is a joint collaboration between EURORDIS (European Rare Disease Organisation) and NORD (National Organization for Rare Disorders).
Living with a genetic or rare disease can impact the daily lives of patients and families. These resources can help families navigate various aspects of living with a rare disease.
Financial Resources
The HealthWell Foundation provides financial assistance for underinsured patients living with chronic and life-altering conditions. They offer help with drug copayments, deductibles, and health insurance premiums for patients with specific diseases. The disease fund status can change over time, so you may need to check back if funds are not currently available.
These resources provide more information about this condition or associated symptoms. The in-depth resources contain medical and scientific language that may be hard to understand. You may want to review these resources with a medical professional.
Where to Start
Genetics Home Reference (GHR) contains information on Variegate porphyria. This website is maintained by the National Library of Medicine.
MedlinePlus was designed by the National Library of Medicine to help you research your health questions, and it provides more information about this topic.
The National Digestive Diseases Information Clearinghouse (NDDIC), part of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), offers information on this condition. Click on the link to view information on this topic.
The National Organization for Rare Disorders (NORD) has a report for patients and families about this condition. NORD is a patient advocacy organization for individuals with rare diseases and the organizations that serve them.
In-Depth Information
Medscape Reference provides information on this topic. You may need to register to view the medical textbook, but registration is free.
The Monarch Initiative brings together data about this condition from humans and other species to help physicians and biomedical researchers. Monarchs tools are designed to make it easier to compare the signs and symptoms (phenotypes) of different diseases and discover common features. This initiative is a collaboration between several academic institutions across the world and is funded by the National Institutes of Health. Visit the website to explore the biology of this condition.
Online Mendelian Inheritance in Man (OMIM) is a catalog of human genes and genetic disorders. Each entry has a summary of related medical articles. It is meant for health care professionals and researchers. OMIM is maintained by Johns Hopkins University School of Medicine.
Orphanet is a European reference portal for information on rare diseases and orphan drugs. Access to this database is free of charge.
PubMed is a searchable database of medical literature and lists journal articles that discuss Variegate porphyria. Click on the link to view a sample search on this topic.
Questions sent to GARD may be posted here if the information could be helpful to others. We remove all identifying information when posting a question to protect your privacy. If you do not want your question posted, please let us know. Submit a new question
I have just had my DNA report returned to find that I am a carrier of variegate porphyia as is my daughter. We both are asymptomatic. I am not South African Dutch. How many second generation and now third generation Americans are diagnosed yearly? If I have never had symptoms (age 65), will I have symptoms?See answer
I come from a Central Oregon town with the population of about 1100 people. I moved back here to Central Oregon, where I was raised after 40 years of being away. I took my grandson to a local ER in Redmond, OR (population about 26,000) after an accident where he hurt his foot which needed to be checked out. I was making small talk with him in the waiting room and told him of some new information about porphyria I had learned. A woman in the waiting room overheard our conversation. So, as it goes in small towns; we started talking. She told me that she had porphyria also.
She was called in to see the triage nurse but her husband remained and we visited. I exchanged contact information with him and we briefly traded stories. She came back in the room shortly. My first question after finding out what kind of porphyria she had (AIP), I asked her how she was diagnosed; as I knew of my own 'war stories' of finally finding the correct diagnosis.
Her story was similar to mine and other porphyria patients, being brushed off by many regular doctors who do/did not understand the cluster of symptoms or level of pain involved during a porphyria attack. If your symptoms don't exactly fit a text book illness model then it is all in your head been-there-done-that. I was diagnosed about a year ago after several Oregon doctors turned their back on me not wanting to deal with the local medical political climate. The few who didn't turn me away hung in there with me and saved my life.
My new friend (from the ER waiting room) was diagnosed about 14 to 16 years ago in Eugene, Oregon. She was deathly ill, hospitalized and was first diagnosed with Guillain-Barre syndrome, yet they still did not know exactly what was going on with her. Her doctors were all puzzled.
She had one doctor who used her personal three days off from work researching the internet on her behalf and wondered if she might have porphyria. This doctor did the porphyria testing and sure enough she nailed the correct diagnosis of AIP. Those kind of doctors are few and far between.
What I find amazing about our chance encounter in the local ER waiting room was that we both found out we were afflicted with this very rare disease in a sparsely populated area. One of us being among only approximately 1 in 100,000 porphyria patients affected in the US. It makes our chance meeting even more amazing
My personal take on just how rare porphyria is: there are probably more porphyria patients out there that could be identified if our doctors had more exposure in their training about the disease; and lab technicians were trained to collect the specimens and process the samples correctly. That is my nickel's worth
#PAW2018 Medical Moments ALAD-Deficiency Porphyria (ADP)
Thursday - April 26, 2018 @ 12:00:03
ALAD-Deficiency Porphyria (ADP)
#PAW2018
What is δ-Aminolevulinic Acid Dehydratase Porphyria?
ADP is a severe disorder caused by a deficiency of the enzyme δ-aminolevulinic acid dehydratase (ALAD) which results in an increase of 5-aminolevulinic acid (ALA) in the liver, other tissues, blood plasma, and urine. In addition, urine coproporphyrin and erythrocyte protoporphyrin are increased. ADP generally presents with sudden attacks of severe stomach pain that last for several days.
Who gets δ-Aminolevulinic Acid Dehydratase Porphyria?
All of the reported cases of ADP have been males, in contrast to the other acute porphyrias. ADP is the least common of all the porphyrias with less than 10 cases documented to date. This is an autosomal recessive disease, whereas the other three acute porphyrias are autosomal dominant. Each parent of an affected individual must have a mutation in one of their ALAD genes and both must pass their mutation on to their child.
What causes δ-Aminolevulinic Acid Dehydratase Porphyria?
ADP is caused by a deficiency of the enzyme δ-aminolevulinic acid dehydratase (ALAD).
How is δ-Aminolevulinic Acid Dehydratase Porphyria (ADP) diagnosed?
There are many laboratory tests available for the porphyrias, and it is often difficult to decide which should be chosen. Many of these tests are expensive and the results are often difficult to interpret. When abdominal and neurological symptoms suggest an acute porphyria, the best screening tests are urinary aminolevulinic acid (ALA) and porphobilinogen (PBG). DNA testing to identify the specific mutation in an individuals porphyria-causing gene is the most specific and sensitive test to confirm the diagnosis of a specific porphyria. Before requesting DNA testing, it is recommended that patients have biochemical testing (urinary, stool and/or plasma porphyrins and porphyrin precursors (ALA and PBG) and/or enzyme assays). However, biochemical testing may be inconclusive.
What are treatments for δ-Aminolevulinic Acid Dehydratase Porphyria?
Treatment is the same as in the other acute porphyrias. For the acute porphyrias, hospitalization is often necessary for acute attacks. Medications for pain, nausea and vomiting, and close observation are generally required with monitoring of salt and water balance. Harmful drugs should be stopped. Attacks are treated with either glucose loading or intravenous administration of hemin (Panhematin). Attacks can be prevented in many cases by avoiding harmful drugs and adverse dietary practices.
Whats Your Shade of Purple?
Thursday - April 26, 2018 @ 07:00:17
Whats Your Shade of Purple?
#PAW2018
Friday April 27th 2018
Show us your Purple APF Spirit!
Look for the pictures in the main open APF FB group!
We will have a winner!
Lets have fun!Show us your Purple! Snap a picture and post it! Look for the OPEN FB (What's your Shade of Purple section)
#PAW2018 Meet Milton Cubas PAW 2018 PAW
Wednesday - April 25, 2018 @ 12:30:31
#PAW2018
MEET MILTON CUBAS, APF Member and ALAD Porphyria (ADP) patient.
My name is Milton Eduardo Cubas and I am 29 years old. Sports and physical activities with others are two of my favorite things.
My current studies at Miami Dade College are to become a math teacher.
I had my first porphyria attack in a very hot summer day of 2000 at a castle in Cartagena, Colombia named Castillo San Felipe de Barajas. I was with family members and I remember feeling fatigued while taking a tour of the castle. I drank a whole bottle of water after leaving. Once I got in a taxi I felt nausea. I felt tired. We stopped to eat something but I would vomit water until nothing would come out. I had gone to hospitals in Colombia and they couldnt find anything. I remember feeling a lot of pain while looking for anyone to tell me what is going on. I had to cut my trip to Colombia short because we couldnt find anyone to help me and my pain was getting worse.
I went to Miami Childrens Hospital and my attack got better and the doctors still couldnt find anything. I would have an attack every month or two for a year until a doctor named Elsa Vasconsuelos told me that I may have Porphyria. Soon after that, my dad found Dr. Anderson. I would take two or three trips to Galveston, Texas until he diagnosed me in February 2003 with aminolevulinic acid dehydratase-deficient porphyria, which is abbreviated as ADP.
Through my teenage years, I had attacks nearly every month. Once I hit my 20s the attacks spread out more. The longest Ive spent without an attack is two and a half years but after that I had 3 attacks really close to each other that affected my neuropathy a lot. My last attack was mid-July of 2017.
I get hematin every single Friday. I have tried spacing out the hematin to every 10 days or every 14 days but I would get an attack after that. During the attack, I would have to be hospitalized to get hematin for four days every day. I get heme in an infusion center. Some of my negative experiences are that some nurses dont know how to administer the hematin the correct way when Im hospitalized.
The neuropathy has affected me every day from dressing myself to doing homework. I have trouble with extending my fingers that stops me from doing hand gestures. I have foot drop where I could lose my balance easily. It has forced me to try find anywhere where there is AC.
I could last longer in the sun now but in the summer I cant. There have been positive experiences, too. I have met a lot of wonderful people, nurses and doctors, who have helped me when Im trying to get better.
My Family has supported me and helped me whenever I couldnt drive or when I was hospitalized. I want to become a Math teacher and I am studying really hard to graduate. I want to get my hands better.
For the people who have Porphyria: Knowledge is Power.
The more you know about Porphyria the stronger you will be.
It is also important to participate in research and I am in the longitudinal study at UTMB.
If you are interested in learning more about the Acute Porphyrias please visit porphyriafoundation.org Please visit us by calling Edrin @ 1/866/APF/3635 to get a free patient packet or a comprehensive Dr. Kit for your medical professionals
Congenital erythropoietic porphyria (CEP) is the rarest type of porphyria and is commonly seen in infancy.[1] It is characterized by severe skin photosensitivity that may lead to scarring, blistering, and increased hair growth at the face and back of the hands.[2][3] Photosensitivity and infection may cause the loss of fingers and facial features.[1] Symptoms of CEP range from mild to severe and may include excessive hair growth throughout the body (hypertrichosis), reddish discoloration of the teeth, anemia, and reddish-colored urine.[4] In CEP, there is a defect in the synthesis of heme within the red blood cells of bone marrow.[3][4] This defect leads to an increase in the buildup and, therefore, waste of porphyrin and its precursors, which leads to the signs and symptoms.[3] Inheritance is autosomal recessive. It is caused by mutations in the UROS gene.[3] Treatment for CEP may include a bone marrow transplant and hematopoietic stem cell cord blood transplantation.[2][1]Blood transfusions or spleen removal may also reduce the amount of porphyrin produced by the bone marrow. Affected people must avoid sunlight exposure.[1]
This table lists symptoms that people with this disease may have. For most diseases, symptoms will vary from person to person. People with the same disease may not have all the symptoms listed. This information comes from a database called the Human Phenotype Ontology (HPO) . The HPO collects information on symptoms that have been described in medical resources. The HPO is updated regularly. Use the HPO ID to access more in-depth information about a symptom.
The resources below provide information about treatment options for this condition. If you have questions about which treatment is right for you, talk to your healthcare professional.
Management Guidelines
The American Porphyria Foundation offers a document that includes information about porphyria, types, testing, and treatment with Panhematin. Click the "document" link above to view these guidelines.
#PAW 2018 The Real Side of Living with CEP ~ Justin Hamilton
Tuesday - April 24, 2018 @ 12:30:00
#PAW2018 Justin Hamilton
Congenital Erythropoietic Porphyria (CEP)
Thank you Justin for participating in this interview.
(Congenital Erythropoietic Porphyria) (CEP))
We take a close look at a day in the life of Justins Life with CEP, well also learn what it is and just how rare it really is.
So at what age did you start experiencing problems? What were they?
When I was first born. They noticed in the hospital my urine was a red fluorescent color. They thought it had something to do with my kidneys.
Did anyone else in your family have a history of CEP?
No
When and how were you diagnosed? Where?
When I was 6 months old. They did blood test and cultures. I was diagnosed at the McCook, Nebraska Clinic. (Currently 150 or less reported cases)
Did they get your diagnosis right the first time?
Yes, besides thinking it was my kidneys. Mom then noticed blisters when the sun would be on me. They then did test to find out I had CEP.
How do you feel about having CEP the ups and downs?
CEP sucks, I wouldn't wish it on my worst enemy if I had one. The ups to CEP there are not really any at all, other than that it gives you a look at life to strive to be positive, making you a fighter for the things that most people take for granted. The downs are endless and new every day. The fact that you are unable to go outside without being all covered up and drenched in sunscreen. The pain and the scarring that happens from any tiny bit of exposure. Not being able to be "normal" like others that can run around outside and soak up the sun. Growing up and living with CEP people staring at you and giving you mean looks because they are not aware of what really is going on. There are times that Im down and stressful times. Trying to cope with all that is thrown at you is a daily struggle.
What are your symptoms?
How have you managed your symptoms? Symptoms are any exposure to the sun or certain lights I get blisters, some big some small. All very painful, and then after they pop they take weeks to heal. Then when they do heal they leave scars that limit normal skin growth. Pain becomes a normal thing that I become used too and just pray it helps fast each time. Trying to manage it is just lots of sunscreen and coverage when in the sun. I believe everything happens for a reason. So I just take what the good lord gave me and make the best of it.
Have you learned anything from having a rare disease?
CEP has taught me to take this disease seriously. I wish I would of been more protected when I was younger age and not been so stubborn when putting on sunscreen, long sleeved shirts, hats and so on. I seem to learn something new every day.
How have your Doctors treated you in the past and present?
They have treated me with antibiotics for infections from my sores. They have had me use steroid creams for my sores. Really that about it. This disease did attack one of my eyes. Which I now don't have much vision left in. Also caused a small hole in that eye. I see an eye specialist regularly. They put glue in the hole and a contact over to keep it stable. My doctors have been supportive and helpful with everything that comes with it.
What are your future goals? Hobbies that you enjoy?
My goals in life are to live as long as I can be watching my Kids growing up and spending time with my wife. I try to stay as covered as possible. And take each day like it was my last. I enjoy the outdoors which is hard since that is where I get most of my exposure from. But I live raising crops, animals, hunting, and fishing. My love for race cars keeps me going.
Do you work and what do you do?
In try to keep busy. Working hard is always been a big part of my family's way of life. I try my best to continue to help family and friends with what I am limited too. I try to help at the farm or the family car dealership when I can.
Are you Married & do you have children & how has this impacted you in any way?
I am happily married since 2011, since then we have two beautiful children. A boy Brayden who is 2 1/2 and a girl Brylee who is 1 1/2. They are my reason for living. I love then so much. There are some thing that limit me to my husband and father duties. With my hands being so small and sore (my hands are ½ the size of a normal persons due to the severe scaring) its hard for me to change diapers, simple stuff like that. I can't play with them outside on a nice day for a very long period of time like I would like to be able too.
If Holly would like to comment, how do you feel about Justin as a husband and father and how does he handle the CEP daily?
He is an amazing husband and an even more amazing dad. There are some things that limit him. But we are aright with that. I understand and try to help him in every way possible to make things easier on him. He has good days and bad days. He does not shoe to anyone else how much pain he might be in. But when it is just the two of us he is able to open up some more. He is a very positive attitude about it all. Some days he may be in enough pain he stays in bed all day. But the next he is ready to go. The kids and I love him so much.
What encouragement would you give to others that have CEP and why raise awareness?
All I can say is words of encouragement. Don't take life for granted. Take your disease seriously and take the needed precautions no matter how much it sucks. Keep positive and have faith. There will be good and bad days but how you react to it is your choice. So stay positive and live life to the fullest. It is very important to spread awareness so that others may understand. The more people and doctors that know more about it they can understand to make it easier on me and ways to help.
If the APF has helped you at any given point, how have they helped you?
The APF has helped out greatly. The thing I like the most is it brings all fellow porphyrians together to tell their stories and help one another to make this disease better to live with. I am very thankful and blessed even with CEP.
#PAW 2018 Medical Moment ~ Porphyria cutanea tarda (PCT)
Tuesday - April 24, 2018 @ 07:00:00
#PAW2018 Porphyria cutanea tarda (PCT) is a form of porphyria that primarily affects the skin. People affected by this condition generally experience "photosensitivity," which causes painful, blistering lesions to develop on sun-exposed areas of the skin (i.e. the hands and face). Skin in these areas may also be particularly fragile with blistering and/or peeling after minor trauma. In some cases, increased hair growth as well as darkening and thickening of the affected skin may occur. Liver abnormalities may develop in some people with the condition and PCT, in general, is associated with an increased risk of liver cirrhosis and liver cancer.[1][2] In most cases, PCT is a complex or multifactorial condition that is likely associated with the effects of multiple genes in combination with lifestyle and environmental factors. For example, factors such as excess iron, alcohol, estrogens, smoking, chronic hepatitis C, HIV and mutations in the HFE gene (which is associated with the disease hemochromatosis) can all contribute to the development of PCT.[3][1] Less commonly, PCT can run in families (called familial PCT). Familial PCT is caused by changes (mutations) in the UROD gene and is inheritedin an autosomal dominant manner.[4][1] Treatment may include regular phlebotomies (removing a prescribed amount of blood from a vein), certain medications, and/or removal of factors that may trigger the disease.[1][2]
This table lists symptoms that people with this disease may have. For most diseases, symptoms will vary from person to person. People with the same disease may not have all the symptoms listed. This information comes from a database called the Human Phenotype Ontology (HPO) . The HPO collects information on symptoms that have been described in medical resources. The HPO is updated regularly. Use the HPO ID to access more in-depth information about a symptom.
The resources below provide information about treatment options for this condition. If you have questions about which treatment is right for you, talk to your healthcare professional.
Management Guidelines
Orphanet Emergency Guidelines is an article which is expert-authored and peer-reviewed that is intended to guide health care professionals in emergency situations involving this condition.
The American Porphyria Foundation offers a document that includes information about porphyria, types, testing, and treatment with Panhematin. Click the "document" link above to view these guidelines.
If you need medical advice, you can look for doctors or other healthcare professionals who have experience with this disease. You may find these specialists through advocacy organizations, clinical trials, or articles published in medical journals. You may also want to contact a university or tertiary medical center in your area, because these centers tend to see more complex cases and have the latest technology and treatments.
If you cant find a specialist in your local area, try contacting national or international specialists. They may be able to refer you to someone they know through conferences or research efforts. Some specialists may be willing to consult with you or your local doctors over the phone or by email if you can't travel to them for care.
You can find more tips in our guide, How to Find a Disease Specialist. We also encourage you to explore the rest of this page to find resources that can help you find specialists.
Research helps us better understand diseases and can lead to advances in diagnosis and treatment. This section provides resources to help you learn about medical research and ways to get involved.
Clinical Research Resources
ClinicalTrials.gov lists trials that are related to Porphyria cutanea tarda. Click on the link to go to ClinicalTrials.gov to read descriptions of these studies.
Please note: Studies listed on the ClinicalTrials.gov website are listed for informational purposes only; being listed does not reflect an endorsement by GARD or the NIH. We strongly recommend that you talk with a trusted healthcare provider before choosing to participate in any clinical study.
Patient Registry
The Porphyrias Consortium is a team of doctors, nurses, research coordinators, and research labs throughout the U.S., working together to improve the lives of people with this condition through research. The Porphyrias Consortium has a registry for patients who wish to be contacted about clinical research opportunities.
Darlene is 44 years old, married, with two children. She lives on beautiful eastern Long Island, New York, which pokes out past the Long Island Sound and towards the Atlantic Ocean. Living out there, having two children to look out for, and working as teaching assistant in a local school Darlene has plenty of opportunities to be exposed to the sun.
Exposure to the sun eventually marked Darlene's symptoms unambiguously last Spring, but they started before that, with tea-colored urine, prompting her primary care physician to send her to a rheumatologist to be evaluated for possible lupus. While she was seeing the rheumatologist, her primary care physician became concerned about her gallbladder. It was in the process of getting ready to have her gallbladder removed that Darlene started getting a lot more exposure to the sun.
Wanting to lose a little weight and get in better physical condition, she started spending more time walking out of doors. This was in May of 2008, and that was when her blisters started forming prickly bumps on the backs of her hands that turned into half-dollar sized blisters that refused to heal. The blisters were very painful and getting worse, prompting her doctors to consider some sort of sun allergy or other allergic reaction.
Finally, Darlene visited an urgent care clinic, where doctors did a smear of the blisters and sent her to a dermatologist, who did a skin biopsy, and diagnosed her with Porphyria Cutanea Tarda (PCT). Her age makes Darlene typical of PCT patients the disease gets the name tarda (Latin for "late") because patients usually do not experience symptoms before they are in their 40s.
Once she had a diagnosis of Porphyria, Darlene found out about the Mount Sinai Porphyria Clinic (in New York City, three hours from her home) through a friend of her aunt who had heard of Dr. Lawrence Liu, one of two doctors who staffs the clinic. She describes meeting Dr. Liu for the first time as an almost revelatory experience, calling him "a breath of fresh air," for his compassionate bedside manner, and for being the first doctor she encountered who really understood Porphyria and took an interest in all of its effects on her. Dr. Liu was also the person who let Darlene know about the existence of the APF.
Since then, Darlene has become an APF member and started receiving the newsletter, and has carefully followed Dr. Liu's advice to avoid the sun while her blisters heal, and to avoid alcohol entirely. Her local doctor was unfamiliar with treatments beyond phlebotomy for PCT, and did not want to remove any of her blood as her counts were already low. Dr. Liu started her on low-dose chloroquine (taken in pill form), which was improving her symptoms somewhat when we spoke. Dr. Liu also did a biopsy of her liver and found out that she fortunately suffers none of the liver damage that sometimes affects PCT patients. The liver biopsy was something she had asked her local doctors about with no reaction.
In addition to the blisters, Darlene had the additional hair growth on her face and legs that is characteristic of PCT. But she says the worst part is having blisters on her hands, because "you do everything with your hands," making it easy to reinjure them, and hard for them to heal. Working with children, and not wanting to make waves at work or air her problems in public, Darlene tried strenuously to protect herself from the sun while doing playground duty while the kids were at recess. She wore gloves and big hats, and tried to watch the kids from the shady portions of the schoolyard. She is lucky enough to have a school principal who asked about her out of concern and has now adjusted her responsibilities so that she can stay out of the sun.
On her way to diagnosis, Darlene had a surgery she now believes could have been avoided with proper treatment of her PCT, and in addition to suffering symptoms of the disease itself, she has lived with the stress and grief of symptoms her doctors would not recognize. In this new, hopeful period in her life, now that her care is being guided by an expert, she speaks about having newly increased sympathy for the difficulties that elderly and infirm people suffer. It is our hope that with appropriate treatment and precautions, Darlene will achieve a complete recovery from her own illness, and that her newfound sympathy for others will no longer have to be accompanied by her own suffering!
#PAW2018 We Go One on One with Skyler Paris & EPP
Monday - April 23, 2018 @ 11:30:11
#PAW2018
Name: Skyler Paris Age: 17
â?¢ How old were you when you were diagnosed?
5years old.
â?¢ Do you remember your first flare/reaction?
Yes, I was two years old at the Beach in Florida
â?¢ What did it feel like to you?
It was hard for me to remember, I remember the pain & bath I had made it worse
*The liver failure/transplant caused a rare porphyria side effect of nephropathy that left me paralyzed and on mechanical ventilation. It has taken nearly a year to get to where I am now. Doctors were only able to find a few documented cases of this occurring in porphyria patients.
â?¢ What things help you feel better? (Cool water, ice, shade, bath, clothes)
Sleep
â?¢ How long does it take before you start to feel better?
2-5 Days before I start to get relief.It really varies for me.
â?¢ What kind of clothing/trends do you wear when you go outside or in bad lighting?
Lots of long sleeved shirts, face shields, ball cap, sunglasses, and gloves
â?¢ What is your most favorite sport to play or watch? Do you play it inside or outside? During the day or at night?
Started with indoor activities when younger (bowling, jiu-jitsu) but hard to keep up with just because of travel. Easy to get burned out on certain activities when they are so limited. Will start going to Nashville Predator hockey games when Im recovered. SEC football is my favorite (GO VOLS)
â?¢ What ways are you able to adapt to do certain activities outside?
Trying to go outside around sunset when the sun is low, while keeping covered up.
â?¢ What fun things are you able to do inside your home while the sun is out?
Video Games, Cooking, having friends over
â?¢ What has been the best vacation ever?
Camping with my best friend
â?¢ Have you met any other kids with EPP before?
I did have someone come see me in the hospital, but I had just had my liver transplant and was not responsive. I was on a ventilator and very sick. I dont remember, but it was very nice for my mom to meet someone.
â?¢ Do you go to School/Homeschool?
Before complications with my liver and needing a transplant, I went to regular school.
â?¢ Do you have to go by car, bus or walk? I drove myself
â?¢ What things do you have to do to protect your skin?
Cover with long sleeves, gloves, hat, face shield, and sunglasses. Prior to last April the indoor lights didn't bother me. I now have to be covered under fluorescent lights but have not been back to school or left the hospital since then. They have changed lights in my room to LED.
â?¢ Is it hard for you to tell your friends, teachers or family what EPP is?
As Im older it is easier to explain. When I was young it was difficult to get the schools to believe me even with doctor notes. My mom had to be very firm and direct with school administration when they didn't believe me. She brought them pictures of what I looked like during a reaction.
â?¢ What do you tell them?
Basically, that the sun, even when its overcast, sets off a photo toxic reaction in my blood. I let them know that it starts to hurt before you see any symptoms.
â?¢ What things do you do if you feel sad or left out?
Never really felt left out. Didn't let it bother me.
â?¢ What do you want to be/do when you grow up?
Mechanical Engineer
â?¢ What advice can you share to help other kids that have EPP?
Speak up and reach out to others. The technology we have today makes this possible.
â?¢ Parents- How do your children deal with the pain/reaction/flare?
He goes into a dark room and trys to distract himself. We give Medications sometimes for flares, but it may act as a placebo more than anything. Additional Medications taken daily has helped the flairs not be as bad.
â?¢ Would you benefit from a new treatment?
Had this been available previously it would have been life changing! Even at this time when a bone marrow transplant will cure him, it would have been easier for the hospital to care for him without having to worry about lights. Even trips to the ICU were delayed while the lights were changed out.
Skyler, Thank you for your words of wisdom!Keep living each day to the fullest. Know your surrounded by a loving family and friends and now that youre in another medical facility I hope that each day you gain the strength and determination to follow your dreams.
#PAW2018 Medical Moments on EPP & XLP
Monday - April 23, 2018 @ 07:00:11
#PAW2018
Important Considerations
Consult a specialist. Because EPP is a rare condition, most physicians are not knowledgeable about it. Primary care or Emergency Room doctors can contact EPP expert: Dr. Micheline Mathews-Roth, Harvard Medical School, 617-525-8249. The American Porphyria Foundation, 713-266-9617 can also provide further information.
Alert the anesthesiologist to use an anesthetic that doesnt block bile flow.
Be careful with medication. Avoid damage to the liver. Avoid griseofulvin, estrogen or any drugs that block bile flow.
Be careful with surgical lights. Strong operating room lights can cause photosensitivity in internal organs. Cover lights with clear window film, such as CL5-200-X from Madico Co. Keep skin covered with surgical drapes and keep exposed internal organs as covered as possible. It is extremely important to shield the surgical lights during abdominal surgery. The approach for superficial surgeries is to shield the skin as much as possible, and to work quickly. In an emergency situation, where better shielding materials are not available, use as much cloth as possible to shield the site.
Be careful with lights. Problems with fluorescent lights are common. Incandescent bulbs are generally acceptable. However, companies may be changing the filaments and thus varying output.
The risks from surgical lights, anesthetics and drugs are great enough that all EPP patients should wear Medic Alert bracelets, even those who have mild enough cases that they can lead quite normal lives with just a few adjustments.
Drugs
Vitamins: Taking Vitamins A and D in excess can cause major health problems. Taking beta carotene, even in large doses as for EPP, does not lead to Vitamin A toxicity, because the body stops splitting beta-carotene into Vitamin A when it does not need the Vitamin A any more. Vitamins E and K can also be toxic at several times the recommended dose.
Celebrex: It has been reported that Celebrex, a non-steroidal anti-inflammatory drug (NSAID), helped reduce photosensitivity. This has not been proven. NSAIDs are powerful drugs and can cause very severe complications, such as stomach and esophageal ulcers and hemorrhages, and some liver problems. Thus, if you need to take NSAIDs for arthritis and other conditions and you find that they happen to decrease your photosensitivity, it is important to ask your doctor to monitor your liver chemistries and porphyrin levels every 4-6 months.
Laser Treatments
Laser treatments for hair removal or eye surgery have been a subject for question. According to Dr. Roth, laser treatments have not been a problem for EPP people, as long as they check with their doctor as to the output of the laser. It should not be between 400 and 650 nanometers. In addition, if the person is having hair removal treatment, the doctor should irradiate a small area of the skin to be treated for the length of time it will take to do the hair removal to ascertain if the patient will get any EPP symptoms from the laser radiation. It is important to wait for as long as it usually takes your symptoms to develop following sun exposure.
Dietary Considerations
There have been a number of reports to suggest that dietary fish oils seem to lessen the symptoms of EPP. At this point, none of these claims can be validated until careful studies under medical supervision are conducted and FDA approval is obtained. This would be a treatment, not a cure. The oils may work by acting as anti-oxidants. One major brand of fish oils is Maxepa, distributed by Twin Labs. EPP people sometimes have low hemoglobin. Therefore, it is suggested that you have enough iron in your diet to meet the recommended dietary allowance (RDA) for this nutrient to avoid becoming anemic. The RDA is 10 mg/day for men, 15 mg/day for women 25-50 years, and 10 mg/day for women over 50. For pregnant women, the suggested allowance is 30 mg/day. It is important to check food labels for iron content.
Women and Osteoporosis
There are several methods of treating osteoporosis, including increasing calcium intake, exercise, hormone (estrogen/progesterone) replacement therapy in postmenopausal women, testosterone therapy for certain men, and calcitonin. There are some experimental drugs being studied at this time, but they have not yet been approved by the FDA.
For women with EPP, hormone replacement therapy (HRT) can be used if you feel that the benefits outweigh the risks, according to our liver consultant Dr. Joseph Bloomer. The risk is that these hormones may impair protoporphyrin excretion from the liver and thus can lead to liver problems (the reason that EPP people are discouraged from taking birth-control pills). If you do decide to take HRT, it is crucial to follow closely your protoporphyrin levels and your liver chemistry blood tests (at least twice a year). If there is any increase in either liver chemistry or protoporphyrin levels, HRT should be stopped immediately. Each case must be judged on an individual basis, so do discuss this with your personal physician. For men, osteoporosis is sometimes treated by giving testosteronethis hormone also affects the liver in the same way that estrogen does, thus the same cautions apply.
Youngsters with EPP
Camp Discovery
Camp Discovery offers a wonderful summer camping experience for young people with skin disorders like Porphyria.
Every year, the American Academy of Dermatology sponsors a week of fishing, boating, swimming, water skiing, arts and crafts, and just plain fun.
Under the expert care of dermatologists and nurses, Camp Discovery offers the opportunity to spend a week with other young people who have similar skin conditions. Many of the counselors have serious skin conditions as well, and can provide support and advice to campers.
There is no fee for camp. Full scholarships, including transportation, are provided by the American Academy of Dermatology through generous donations of their members and other organizations. Members of the Academy are asked to recommend candidates for Camp Discovery, so ask your child's doctor about sending your child to Camp Discovery.
Disneyland and Disney World are responsive to people with sun sensitivity. They will provide a pass to enable you to enter attractions without waiting in line in the sun.
Disneyland Go to "City Hall" and explain your problem with photosensitivity. You should bring a physician's letter with you as well as an APF brochure explaining the type of Porphyria you have.
Disney World Proceed to the "Guest Relations" office at any park (Magic Kingdom, EPCOT, etc.) and request the Special Assistance Pass.
Remember to bring a doctor's note and explanation of your condition, because it is not necessarily visible. People on duty may not be familiar with light sensitivity and its consequences.
#PAW2018 Short story on Louise Coomber EPP
Sunday - April 22, 2018 @ 12:30:03
#PAW2018
Louise Coomber
Type of Porphyria:
Erythropoietic Protoporphyria (EPP)
I had my first symptoms of EPP at 18 months old but didn't get a diagnosis until the age of 31! I was repeatedly told by doctors through my teenage years to just stay out of the sun. We just don't have widespread knowledge of EPP within our health system in the UK. I eventually became obsessed with finding out what was wrong with me, so I went to my doctors with a stack of information about EPP, and finally found my answers. It was the most incredible feeling after all those years of feeling different but not knowing why. Also meeting and talking to other people who have EPP is an incredible feeling. I have never had a reaction in the winter, with the exception of skiing in Italy, which I put down to the intensity of the sun at altitude. I'm lucky in the sense that in England we only have 5-6 months of warm enough weather to affect me. I also have a daughter who is 9 and doesn't have EPP. It is a struggle in the summer. I have suffered so many feelings of guilt through her life when I have had to say 'Mummy just can't do it.' We tend to holiday outside of season and mainly take advantage of our beautiful coastline. However, shaking the feeling of guilt that she doesn't get to experience the traditional warm, 'beach holiday'' is a struggle. My family is amazing, and my daughter is lovely and protective now that she understands my EPP.
#PAW2018 Medical Moment What is HCP
Sunday - April 22, 2018 @ 07:00:01
#PAW2018
What is Hereditary Coproporphyria?
HCP is due to a mutation in coproporphyinogen oxidase (CPOX), which is part of the pathway that produces porphyrins and heme. It is an autosomal dominant disorder, meaning that a mutation is present in only one of the pair of CPOX genes. The incidence of active HCP appears to be at most 2 per 1,000,000. The prevalence of the genetic carrier state is unknown.
Who gets Hereditary Coproporphyria?
HCP is termed a disease with low penetrance, meaning that many genetic carriers (defined by having a CPOX mutation) never have signs or symptoms of active porphyria. Active disease in general requires the presence of environmental factors such as certain drugs, hormones, and dietary changes, as in AIP. Lists are available of drugs that are risky for HCP genetic carriers as well as drugs that are safe (http://www.porphyriafoundation.com/testing-and-treatment/drug-safety-in-acute-porphyria). The worst offenders are barbiturates, sulfonamide antibiotics, anti-seizure drugs, rifampin, and oral contraceptives (progesterone, in particular). Attacks in women may occur after ovulation and during the last part of the menstrual cycle when progesterone levels are high. Reduced food intake, often in an effort to lose weight, as well as infections, surgery, and stressful situations may also precipitate attacks. Alcohol has been implicated in some attacks. People with repeated attacks are at risk for developing chronic renal disease and liver cancer (hepatocellular carcinoma)
How is Hereditary Coproporphyria diagnosed?
The initial test for people with symptoms is quantitative urinary aminolevulinic acid (ALA), porphobilinogen (PBG) and porphyrins. Elevation of ALA, PBG and coproporphyrin (predominantly isomer III) is highly suggestive of HCP. For asymptomatic individuals, the urine studies may be normal, but a fecal porphyrin analysis will show elevation of coproporphyrin III. Screening tests of this kind should be confirmed by DNA analysis to confirm a CPOX mutation.
What are treatments for Hereditary Coproporphyria?
Treatment, complications, and preventive measures are the same as in AIP. Hospitalization is often necessary for acute attacks. Medications for pain, nausea, and vomiting and close observation are generally required. During treatment of an attack, attention should be given to sodium (salt) and water balance. Harmful drugs should be stopped. Attacks are treated with either glucose loading or hemin (Panhematin, Recordati). These are specific treatments that lower the production of heme pathway intermediates by the liver. Glucose or other carbohydrates are given by mouth if possible, otherwise by vein. However, unless an attack is mild, it is now common practice to give hemin as soon as it is available, because it works more quickly than glucose loading, preventing the neurological complications of prolonged attacks.
Patients with severe renal disease tolerate hemodialysis or kidney transplantation. Liver transplantation has been very effective for patients who have repeated attacks and who are resistant to other treatments. However, experience with transplantation as a treatment is still limited.
What is the long-term outlook after an attack of Hereditary Coproporphyria?
The prognosis is usually good if the disease is recognized and treated promptly, before nerve damage develops. Although symptoms usually resolve after an attack, recovery of neuromuscular function (in a severe case) may require several months or longer. Mental symptoms may occur during attacks but are not chronic. Premenstrual attacks often resolve quickly with the onset of menses.
Can attacks be prevented?
Yes, particularly with regard to drugs and diet. Genetic HCP carriers should become informed on drugs and other factors that can lead to symptoms (see above). They should be prepared to point their healthcare providers to drugs and medications to avoid. The American Porphyria Foundation offers a mobile phone app that pulls up this information on line (http://porphyriadrugs.com/). A Medic Alert bracelet is useful for a situation in which the patient is incapacitated. Very frequent premenstrual attacks can be prevented by a gonadotropin-releasing hormone (GnRH) analogue administered with expert guidance. In selected cases, frequent noncyclic attacks can be prevented by once- or twice-weekly infusions of hemin.
Individuals who are prone to attacks should consume a normal balanced diet. Despite on-line discussion, there is no evidence that pushing carbohydrate prevents attacks, and it has the side effect of weight gain, which is undesirable for most people. Fasting, fad diets (for example, high protein) and gastric reduction surgery should be avoided. If weight loss is desired, it is advisable to consult a physician and a dietitian about an individualized diet with modest caloric restriction (ca. 10%), which will produce gradual weight loss without increasing the risk of an attack of porphyria. Exercise is safe in porphyria, and recommended.
#PAW2018 Nathan Wayne Carr ~ How Porphyria can get one feeling low HCP
Saturday - April 21, 2018 @ 12:30:31
#PAW2018
Nathan Wayne Carr ~ How Porphyria can get one feeling low
Type of Porphyria:
Hereditary Coproporphyria (HCP)
How Porphyria can get one feeling low
Recently, I posted on Facebook how I have been dealing with my mental state. I just need to reach out to my Porphyria family. For the past few months, I have not been answering my telephone or keeping medical appointments. Friends have been calling and the power on the cell has been off. I have been feeling overwhelmed, seems as if I only have enough energy to deal with me. My doctor asked me to see psychologist months ago, but I never went to the appointment. I told myself I could handle it. I definitely needed a Prozac adjustment. I am trying hard to climb out of this space, I keep a lot to myself about how I feel, but I can share with you all easily. I need to be true to myself.
When I was asked to write this article regarding depression and anxiety, my mind went back over twenty years ago, when I finally received a diagnosis. I was given information to contact the American Porphyria Foundation, and I was happily waiting to receive the information packet. When it arrived, I was set and ready to read the Porphyria booklet from cover to cover. I started reading, but every time I got to the symptoms section I kept closing the book. I kept getting stuck on mental afflictions, but thought they do not apply to me.
It is morning, and I am awake. I feel as though I arise into a world that is not my own. A world formed from the elements of the earth that my body cannot withstand: the sun, UVA and UVB rays, weeds, trees, mold, grass and a variety of foods. I was once meeting with a doctor and he told me I was in denial of my condition, which was true at the time. I just wanted it to be a bad dream, and I would awake lying on the beach soaking up the sun.
But this is my reality and I too have to except it. I was in a dark place. I felt as though I was in a hole unable to crawl out. I have the support of many wonderful friends and family members that are concerned about me, but, during those times of depression I shut off mostly everyone. I didnt answer my telephone nor returned calls to those who cared.
I had to find a way to pull myself up out of the self-pity I was wallowing in. I was so tired of the pain, nausea, no appetite, weight loss and the constant medical bills that kept pouring in. One day, with the help of friends, I pulled through. I had to remind myself that this too shall pass. I have been here before and made it. I was focusing on what maybe yet to come and not on what was taking place in the moment; that I am surrounded by a loving family and friends who love me as I am and concerned about my well-being. And that God has always been my rock, provider and protector. I am learning to be content.
I also had to make a decision to take better care of myself by keeping my medical appointments that were regularly scheduled. Yes, I do get tired of going to the doctors. I recently joined the YMCA so that I can strengthen my body and clear my mind. At times, workouts can be painful especially during times of muscle weakness or mild crisis. But with a new determination I arise. I thank God for my parents, sisters, daughters, granddaughters and friends that are understanding and encourage me to take better care of myself regardless of my situation.
I have made an appointment with a psychologist. This time I do plan on building a relationship with this individual to help me cope with these issues and find things to do that I enjoy. I do not know why I feel so selfish when I have to make decisions for myself and my well-being regardless of how others feel. I do think that the medical issues I had last year changed me and my body.
I am satisfied with seeing my new psychologist, being able to speak freely about how I feel and to help me cope with daily living. We know that living with Porphyria is very painful and difficult to carry out everything we have planned. I have just learned to expect the unexpected and that I have to be prepared for any changes in my life. From what I have gone through in the past reminds me that I can cope with the future. I am making it one day at a time.
I had to remove some individuals from my life that just could not understand my illness or caused me stress and it made me feel better. I am spending time concentrating on myself, eating more, walking daily, spending time in the gym and surrounding myself with friends and family that make me laugh and do not try to fix me.
I love myself just the way I am. I was beautifully made by the hands of God. He knew me while I was in my mothers womb. So he was aware of my situation and is prepared to help me through it.
Nathan Carr 2/13/2014
#PAW2018 Medical Moment ~ Acute Intermittent Porphyria
Saturday - April 21, 2018 @ 07:00:07
#PAW2018 This is one of the hereditary hepatic porphyrias. Its inheritance is autosomal dominant. The deficient enzyme is porphobilinogen deaminase (PBGD), also known as hydroxymethylbilane synthase (HMB synthase). This enzyme was formerly known as uroporphyrinogen I-synthase, and this term is still used by some clinical laboratories. A deficiency of PBGD is not sufficient by itself to produce AIP, and other activating factors must also be present. These include hormones, drugs and dietary changes. Sometimes, activating factors cannot be identified.
Most people who inherit the gene for AIP never develop symptoms. However, experts recommend that all relatives of someone with AIP obtain testing, to determine who has the genetic trait and who does not. Those who test positive for the trait should be educated as to measures that will help avoid attacks. Prevention is essential to good management.
AIP manifests after puberty, especially in women (due to hormonal influences). Symptoms usually come as discrete attacks that develop over two or more days. Abdominal pain, which is associated with nausea, can be severe and occurs in most cases.
Other symptoms may include:
·nausea
·vomiting
·constipation
·pain in the back, arms and legs
·muscle weakness (due to effects on nerves supplying the muscles)
·urinary retention
·palpitation (due to a rapid heart rate and often accompanied by increased blood pressure)
·confusion, hallucinations and seizures
Sometimes the level of salt (sodium and chloride) in the blood decreases markedly and contributes to some of these symptoms. The skin is not affected.
Diagnosis
Because this disease is rare and can mimic a host of other more common conditions, its presence is often not suspected. On the other hand, the diagnosis of AIP and other types of porphyria is sometimes made incorrectly in patients who do not have porphyria at all, particularly if laboratory tests are improperly done or misinterpreted. The finding of increased levels of delta-aminolevulinic acid (ALA) and porphobilinogen (PBG) in urine establishes that one of the acute porphyrias is present. If PBGD is deficient in normal red blood cells, the diagnosis of AIP is established. However, measuring PBGD in red blood cells should not be relied upon by itself to exclude AIP in a sick patient, because the enzyme is not deficient in red blood cells of all AIP patients.
If it is known that someone in a family has AIP, and their enzyme value is low in red blood cells, other family members who have inherited a deficiency of PBGD can be identified by measuring the enzyme in their red blood cells. Latent cases so identified can avoid agents known to cause attacks. However, in some AIP families, PBGD is normal in red blood cells and is deficient only in the liver and other tissues. Falsely low values sometimes occur due to problems with collecting and transporting the sample.
DNA is the material in cells that encodes all the genetic information of an individual. Many different mutations have been identified in the portion of DNA that comprises the gene for PBGD. However, within a given family, everyone has the same mutation. When that mutation is known for one member, screening of the relatives is straightforward and can be done on DNA from saliva (spit) or a swab of the inside of the cheek. This is now the gold standard of diagnosis and is available through specialty labs. Details are available from the APF or investigators of the national Porphyria Consortium http://rarediseasesnetwork.epi.usf.edu/porphyrias/index.htm.
#PAW2018 Kim Littlewood & AIP~ She lives to tell her experience
Friday - April 20, 2018 @ 21:43:53
#PAW2018 My porphyria was diagnosed in December 2002. I was 4 week pregnant and was put on some medication (I later find out it was on the unsafe list) I asked my midwife if it was safe in pregnancy as I had been (I had been on them 5 days) getting stomach pains she assured me they were safe. The pains got worse and also I started vomiting a lot and my muscles were getting weaker. I visited my GP surgery (I could hardly walk and needed help to walk from the car into the surgery) as I felt really unwell and he took my blood. I went home and went to bed a few hours later the surgery phoned and asked me to go back down to the surgery. As I got out of bed I collapsed and started having a seizure my partner phoned for an ambulance and I was taken to hospital. Luckily for me the Dr on call at A&E had seen a case of porphyria a few months previous in another hospital and tested me.
I was admitted to the hospital over the next few days my pain got worse in my tummy lower back and thighs and I started getting sharp pains (nerve pains) in my hands feet and legs and I couldn't stand. I was still vomiting and was getting fed by a tube. I was diagnosed with a.i.p and the Dr's then did all their research of how to treat me. By this point I couldn't hold my head up my arms up I couldnt support myself to sit up and I couldn't speak. I had virtually lost every muscle in my body even my vocal muscles. I stayed in hospital until May 2003 had speech therapy and physiotherapy but because I kept falling they didn't do to much with me being pregnant. I gave birth to a healthy but tiny little girl in July 2003 and that's when my intense physio started. I learnt to walk again although I still have a lot of muscle damage now as because some of my nerves died during my on going attack there was nothing to stimulate muscle growth.
When my baby was 4 week old I and another attack and was hospitalized again. I had heamatin and glucose and was discharged after a week. I had an attack every few months lasting between 7 -10 days (which required hospital admissions)for about 3 years and then the attacks got closer they came about every 6 week each time requiring admissions to hospital and lasing about 10-14 days. This went on for 2 years. I then had my little boy and when he was about 1 my attacks became every month. My Dr tried switching my hormones off but that didn't stop my attacks .my attacks were always quite bad and needed hospital admissions.
I was also had a weekly infusion of heating via my Hackman line at home between admissions to try and control my attacks. This didn't work either. I was referred to a specialist professor Cox at another hospital who specialized in porphyria and he also tried his best to help me. In Dec 2010 I was admitted again and was there for 10 month as they could not control my attacks they were just getting one under control when another started.
I had another meeting with Professor Cox in Jun 2011 and he suggested he would put me forward for as a liver transplant as I had no quality of life and I was only 31 with 2 children who needed me. So in July they had a meeting and agreed for me to go on the transplant waiting list. I was still an inpatient at this point and in Aug/Sept 2011 my porphyria worsened I went blind which was a side effect of a rare complication to porphyria where my brain was swelling and I was having constant seizures. My family was called in as they didn't think I'd make it through the nightfortunately I did after getting resuscitated about 5 times and been on life support I regained consciousness after a few days and was transferred back to Addenbrooke's Hospital where professor Cox were. Another meeting was held on Fri 8th Sept and they put me to the top of the transplant list.
They came to me on Saturday 9th as they had found a donor but unfortunately after been prepped for theater, they decided they couldn't use the liver as it was too fatty, and I was quite young. The next night, they had another donor, and I was taken to theater and had my transplant through the night. Before I went into theater, my porphyrin levels were 565. The day after my transplant my porphyrin levels had dropped to 2, so they were fantastic and although I had just had a major operation, I felt better than I had felt in 9 years.
It was amazing as I was virtually pain free. I am nearly 5 years post-transplant now and have had no admissions. Yes, I will always have porphyria as it's in my genes so therefore, in all my other organs but because the liver is genetically not mine, I don't have attacks anymore as it was the liver that couldn't handle the porphyria.
Yes I have to take medication for the rest of my life but that is the point I now have a life. It doesn't revolve around admissions. Life is great.
-Kim Littlewood
#PAW2018 PAW April 21-28 2018 Preview
Friday - April 13, 2018 @ 06:00:04
#PAW2018 This year, each day we will bring you a member story of each type, a Medical Fact on each type, Medications approved by the FDA at this time, Research opportunities & how ALL PORPHYRIA members are spreading awareness!
Louise Schlosser BraunI'm speaking to the Emergency Room Department and to several Interns at UCI in Orange and Irvine California. I also have a huge burden this year to reach out to hospitals that are not providing the Pan Hematin treatments. I'm finding several patients experiencing this recently. If you have any particular hospitals that you're aware of, please let me know. I'm doing mailings for those out of state and plan to visit a few locally to spread AWARENESS this year!
Porphyria Awareness Week will be held on April 21-28, 2018. We strive to dedicate this week to promote this group of rare diseases, reduce the stigma associated with porphyria through physician education, and provide support for those affected.
Porphyria Awareness Week is the time for you to bring porphyria awareness to your local communities. We, at the American Porphyria Foundation, encourage You to help raise awareness and provide accurate information about porphyria where you live. The APF will help you accomplish your own activity by providing:
·Porphyria Brochures
·Porphyria Fact Sheets
·PorphyriaLive DVDs
·Information to gain media attention
·PowerPoint presentations,
·Porphyria Awareness week Graphics for Print, Web, and Social media (See downloads below)
·Press releases for local newspapers and television and Much More!
Contact Edrin at the APF office today to request information to be sent to you!
Why I Am Thankful for Being 'Rare' This Rare Disease Day
Wednesday - April 11, 2018 @ 06:00:12
2018
I bet you never thought you would hear me say I am grateful for my rare diseases. Heck, I never thought I would ever say that. Being diagnosed with several rare diseases with no cure is life-altering and a hard concept to grasp at first. Of course, many days are filled with worry, frustration and pain. But today, I am thankful for all of the life lessons my rare diseases have taught me.
1) My rare diseases have taught me patience. Before I became sick, it seemed as if I was always in a rush. Now I realize it is important to slow down and realize life is about a journey. Enjoy this moment.
2) My rare diseases have taught me the importance of friendship. When you become sick, you truly find out who your friends are. My true friends have been there for me through every painful test, hospital admission, and surgery I have endured.
3) My rare diseases have taught me gratitude. I am thankful my rare diseases have made me realize every single breath we take on this Earth is a blessing, a gift not a right. Dont ever take that for granted.
4) My rare diseases have taught me about endurance. I will never stop fighting. I will never give up on my body. My body has made it through every hardship thus far. My body is undefeated against my chronic illnesses.
5) My rare diseases have taught me about optimism. One thing I always remind myself is, It could always be worse. And I truly mean that. I will always know in my heart that things will get better, even on the worst, most painful days.
6) My rare diseases have taught me empathy. My heart breaks for every person struggling with an illness. When I read articles and posts by other spoonies, I can feel in my body how awful their pain is because I have been through it. I have hope better days are coming for each one of you!
7) My rare disease have taught me the meaning of strength. This is a deep strength within myself that I never knew I had. I may not have as much physical strength as I once had before I was sick, but mentally and emotionally, I am strong and my illnesses will not break me.
8) My rare diseases have taught me unconditional love. I never thought at 24 years old I would need help with basic tasks such as walking or showering. But, sometimes I do, because that is the reality of being chronically ill. My fiance has seen me at my absolute worst crying in pain, puking you name it, and he has dealt with it. Never once has he ever complained about helping me. Never once has he or my parents made me feel like a burden. And that, my friends, is the definition of unconditional love.
9) My rare diseases have taught me about faith. I never have been the most religious girl, but getting sick has showed me how important faith is. Not just religion, but faith that things will get better. Faith that everything will be OK in the end.
10) My rare diseases have taught me to appreciate simplicity. I now find joy with the smallest things in life my own bed, instead of a hospital bed, cuddling with my dog, having the energy to get up and cook a meal, and taking a shower without passing out.
So thank you, rare disease, for teaching me how to love this life.
What is Erythropoietic Protoporphyria and X-Linked Protoporphyria
Monday - April 9, 2018 @ 06:00:19
Erythropoietic Protoporphyria and X-Linked Protoporphyria
NORD gratefully acknowledges Manisha Balwani, MD, MS, Associate Professor, Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, for assistance in the preparation of this report.
Synonyms of Erythropoietic Protoporphyria and X-Linked Protoporphyria
EPP
erythrohepatic protoporphyria
protoporphyria
General Discussion
Summary
Erythropoietic protoporphyria (EPP) is a rare inherited metabolic disorder caused by a deficiency of the enzyme ferrochelatase (FECH), which results from changes (mutations) in the FECH gene. Due to abnormally low levels of this enzyme, excessive amounts of protoporphyrin accumulate in the bone marrow, blood plasma, and red blood cells. Some patients with symptoms of EPP have a genetic change in a different gene called ALAS2. When a patient has a genetic change in this gene, the condition is referred to as X-linked protoporphyria (XLP).
The major symptoms of these disorders is severe pain on exposure to sunlight and some types of artificial light, such as fluorescent lights (phototoxicity). On sun exposure, patients may first experience tingling, itching, burning of the skin. After continued exposure to light, the skin may become red and swollen. The hands, arms, and face are the most commonly affected areas. Some people with EPP/XLP may also have complications related to liver and gallbladder function.
Introduction
Erythropoietic protoporphyria is one of a group of disorders known as the porphyrias. The porphyrias are all characterized by abnormally high levels of particular chemicals (porphyrins) in the body due to deficiencies of certain enzymes essential to the synthesis of hemoglobin. There are at least eight types of porphyria. The symptoms associated with the various types of porphyria differ, depending upon the specific enzyme that is deficient. It is important to note that people who have one type of porphyria do not develop any of the other types.
Signs & Symptoms
The most common symptom of erythropoietic protoporphyria and X-linked protoporphyria is severe pain on sun exposure. Some patients may also be sensitive to some types of artificial light. When the skin is exposed to sun, patients first develop tingling, itching, and/or burning of the skin. These symptoms serve as warning signs as longer exposure can result in severe pain. Affected individuals may also have an abnormal accumulation of body fluid under affected areas (edema) and/or persistent redness or inflammation of the skin (erythema). In rare cases, affected areas of the skin may develop sac-like lesions (blisters) and scar if exposure to sunlight is prolonged. However, scarring and/or discoloring of the skin is uncommon and rarely severe. These affected areas of skin may become abnormally thick. The severity and degree of symptoms is different from case to case. Some patients may only be able to tolerate a few minutes of sun exposure while others may be able to tolerate longer sun exposure without symptoms. The amount of sun tolerated may also be different based on weather conditions. Symptoms are often seen during infancy; however, in some cases, it may not occur until adolescence or rarely in adulthood.
In some affected individuals, the flow of bile through the gallbladder and bile ducts (biliary system) may be interrupted (cholestasis) causing gallstones (cholelithiasis) to form. In turn, such stones can cause obstruction and/or inflammation of the gallbladder (cholecystitis). Rarely, affected individuals may also develop liver damage that, in very severe cases, may lead to liver failure requiring transplantation. As liver transplantation does not cure EPP or XLP, a bone marrow transplant following liver transplant may be necessary in some cases.
Symptoms usually start in childhood but diagnosis is often delayed since blistering is not common and, because the porphyrins are insoluble, they cannot be detected on urinanalysis. The diagnosis is made upon finding increased levels of the protoporphyrin in the plasma or red blood cells in both EPP and XLP. Genetic testing is useful to confirm the diagnosis.
Patients with EPP and XLP may also have mild anemia (low blood counts). In many cases, this may be due to low iron stores. They may also have high levels of liver enzymes on blood tests.
Causes
EPP is a rare genetic disorder caused by genetic changes in the FECH gene. The FECHgene is responsible for providing instructions for the body to create an enzyme called ferrochelatase. This enzyme is involved in a long process to make heme, a chemical that functions to transport oxygen around the body. Without enough of the enzyme, ferrochelatase, the body is not able to finish converting a heme precursor called protoporphyrin into heme, causing buildup of protoporphyrins in certain tissues in the body (i.e., the plasma, red blood cells, and the liver). These protoporphyrins also build up in the superficial blood vessels under the skin. These protoporphyrins are highly sensitive to sunlight. When they absorb sunlight, it results in a reaction which causes severe pain and inflammation resulting in symptoms of EPP.
EPP is inherited, or passed down through the generations, in an autosomal recessive manner. Everyone has two copies of the FECH gene, one inherited from the mother and one from the father. Most individuals with EPP have a different gene change on each copy of the FECH genes. On one copy, the change, called a mutation, has stopped this copy of the gene from working properly. On the other copy, there is a small change called a low-expression allele or a polymorphism. This alteration still affects the way the FECH gene works; it produces less ferrochelatase enzyme than normal. This small change is common in the general population, with up to 10% of Caucasians with one copy of this change. This alteration will not cause EPP by itself, and people who have the alteration on each copy of the FECH gene will NOT develop EPP. But when someone inherits the small alteration from one parent and a mutation from the other, they will develop EPP, because there will not be enough enzyme made. Most patients with EPP have the low-expression alteration on one copy of the FECH gene and a mutation on the other copy. The risk for patients with EPP to have a child who also has the condition depends on the genetic changes in their partner.
Some patients with symptoms of EPP have a genetic change in a different gene called ALAS2, a gene located on the X chromosome. When a patient has a genetic change in this gene, the condition is referred to as X-linked protoporphyria (XLP). XLP is passed down through families in an X-linked manner. Males have one X chromosome and one Y chromosome, while females have two X chromosomes.This means that males have only one copy of the ALAS2 gene and females have two copies of the ALAS2 gene. When a male has a mutation is his single copy of ALAS2, he is expected to have symptoms of XLP. In a woman with a mutation in one of her ALAS2 genes, the second functioning copy of the gene can help compensate and may lead to less severe symptoms or no symptoms at all. It is not possible to predict or control the severity of disease in females. Men with XLP pass on their X chromosome to their daughters and their Y chromosome to their sons. Therefore, a man with XLP with pass on his genetic change to all his daughters, and none of his sons. For a female with XLP, she will pass on the X chromosome with the genetic change 50% of the time. Thus, in each pregnancy, there is a 50% chance of having a child with a mutation in ALAS2.
Genetic counseling is recommended for affected individuals and their families.
Affected Populations
EPP is a very rare inherited disorder that affects males and females in equal numbers. It is estimated that the disorder occurs in about 1 in about 75,000 to 1 in 200,000 individuals in Europe. The number of patients affected by these disorders in the US is unknown. XLP accounts for about 10% of cases in the United States. It is more likely to present in males. Females with XLP may or may not have symptoms.
The onset of symptoms affecting the skin usually occurs in infancy, with an average of diagnosis at age 4; however, in some cases, onset may not occur until adolescence or rarely even adulthood.
Related Disorders
Symptoms of the following disorders can be similar to those of EPP and XLP. Comparisons may be useful for a differential diagnosis:
There are several other types of porphyrias, all of which involve deficiencies of specific enzymes. Most of the symptoms of these porphyrias are not similar to the symptoms found in erythropoietic protoporphyria. Individuals with porphyria cutanea tarda and congenital erythropoietic porphyria may develop skin lesions; however, these lesions do not resemble the skin lesions found in EPP. It is important to note that individuals with one type of porphyria do not develop any of the other types. In addition, there are skin disorders characterized by hypersensitivity to artificial light and sunlight besides EPP, such as xeroderma pigmentosum and epidermolysis bullosa. The skin lesions in these disorders do not resemble the skin lesions in EPP. (For more information on these disorders, choose porphyria, xeroderma pigmentosum or epidermolysis bullosa as your search terms in the Rare Disease Database.).
Other skin disorders which may cause photosensitivity include polymorphous light eruption, solar urticaria and drug-induced photosensitivity.
Diagnosis
The diagnosis of EPP and XLP may be made by a thorough clinical evaluation, and specialized laboratory tests. EPP and XLP are usually diagnosed during infancy or early childhood, due to characteristic symptoms, and by testing the red blood cells (erythrocytes) for increased levels of protoporphyrin. Genetic testing is useful to confirm the diagnosis and identify if it is EPP or XLP. This information is useful for genetic counseling and testing family members as both are inherited in a different manner.
Standard Therapies
Treatment
Avoidance of sunlight will be of benefit to individuals with EPP. The use of sun protective clothing such as long sleeves, hats, and sunglasses will also benefit patients. Tanning creams which increase skin pigmentation or sunscreens which contain physical reflecting agents may be beneficial to some patients. Individuals with EPP and XLP may also benefit from window tinting or using films to cover the windows in their car or house. Before tinting or shading car windows, affected individuals should check with their local Registry of Motor Vehicles to ensure that such measures do not violate any local codes.
In EPP, a high potency form of oral beta-carotene (Lumitene, Tishcon) has been used to improve an affected individuals tolerance of sunlight. While some patients report improvement, recent studies show that there is no data to support the benefit of this treatment.
A drug called Scenesse (afamelanotide) has been approved for the treatment of EPP in Europe. This drug is an injectable implant and works by increasing skin pigmentation which provides protection and improves sun tolerance. This drug is not available in the United States and it has not been tested in children.
When iron deficiency is present, iron supplements may be given. A drug called cholestyramine or activated charcoal maybe prescribed to interrupt the circulation of protoporphyrin through the liver and intestines in patients with liver disease.
In addition, individuals with high levels of protoporphyrin in the plasma and red blood cells should be observed closely by a physician for possible liver malfunction that could eventually lead to liver failure.
Liver transplantation has been performed as a life-saving measure in patients with EPP and XLP related liver failure. Bone marrow transplant can also be performed after liver transplant to prevent further damage to the liver.
EPP and XLP patients should take vitamin D supplements as they are likely to have low vitamin D levels since they avoid sunlight. They should also receive vaccination against hepatitis A and B to prevent other causes of liver damage.
Patients should be seen at least yearly to monitor protoporphyrin levels, anemia, liver enzymes, iron and vitamin D levels.
Other treatment is symptomatic and supportive.
Medical Moment by Desiree Lyon
Friday - April 6, 2018 @ 06:30:19
Medical Moment by Desiree Lyon
MEDICAL MOMENT WHAT ELSE CAN IT BE IF IT IS NOT ACUTE PORPHYRIA? Many of you have had negative biochemical and negative DNA results. Please note the list below of possible other diseases that ppl ,who were positive they had acute porphyrias , have discovered they had instead: of porphyria:
Thanks to Mary Schloetter who helped me put together this list of diseases !!! These include disorders from metabolic and mitochondrial diseases: Complex Regional Pain Syndrome, paroxysmal nocturnal hemoglobinuria (Ab and back pain, dark urine, headache, shortness of breath, blood clots), Malonyl-CoA Decarboxylase (Patient is one of about 20 ppl dx'd with this, and first one dx'd genetically), Glucose-6-phosphate dehydrogenase deficiency G6PD (ab and back pain, dark urine, fatigue, dizziness, rapid heart rate, shortness of breath, and fever are triggered by even tiny amounts of soy, citrus fruit, blue dyes, or Vitamin C), MCAD/MCAS, glycogen storage diseases, Wilson's Disease, Mediterranean Fever and CPOX4 (like porphyria but MUST have mercury exposure to set it off), Mast Cell, Lyme disease with more intensive tests, Ehlers Danlos, some liver and kidney diseases and lead poisoning. It is sad also to say that there are thousands of rare metabolic diseases, so hang tight and don't let that stop you seeding a diagnosis.
Certain medications can cause the dark urine often seen in porphyria attacks, namely , anti-malarial drugs, laxatives containing sienna. And , of course the unsafe drugs that precipitate an attacks must be heeded very carefully. Check the Safe/Unsafe list on the APF website whenever you get a new prescription. dont just think the dr knows tha the drug is safe. Many people have been prescribed an unsafe drug, so remind the drs, nurses and dentists. Even certain foods can cause the dark urine seen in acute porphyria, like fava beans aloe, rhubarb.
It is essential to find the correct diagnosis so that you have a chance to be treated or cured. Now that is not easy usually as drs may not be able to diagnose , you may not be able to find the answers. Dont forget this good advice ' THOSE WHO KNOW THE MOST , DO THE BEST!!!!
Personal Story of Arunas Brizgys & AIP
Wednesday - April 4, 2018 @ 06:30:36
Arunas Brizgys
Acute Intermittent Porphyria (AIP)
Greetings to all Sponsors of the Porphyria Club:. I have just recently joined it. My name is Arunas Brizgys (ah-roo-nus birz-geese), male, nearly 55 years old, and I live in Kingwood, Texas. My ethnicity is Lithuanian/Baltic (i.e. east coast of the Baltic Sea, northern Europe).
My path to the club has been a tortuous one, as Im sure it has been for all of you. I also have had hypertension since my mid-20s, which undoubtedly complicated things. My porphyria probably started manifesting itself about 3-4 years ago with intermittent bouts of severe gastritis. At the same time my cardiologist had me on what I now believe to be a too high a dosage of an anti-hypertensive alpha blocker. I was on a series of medications which gave me palpitations, weakness, malaise. I got to spend a night in the ER. So he changed the meds again. Within two weeks my toes started to tingle and I started getting episodes of weakness, like the flu. My cardiologist said it wasnt the meds. So, he upped the dosage. The tingling in my toes became more intense as did the episodes of flu-like weakness, and now plus palpitations and muscle aches. So, over the 4th of July, 2003, I got to stay in the hospital for a day and a half. The doc said there was nothing apparently wrong with me and released me.
During the next few months, the episodes became more frequent and intense. Since my BP was still high, the cardio doc added another medication. Within 3 days I was cramping, nauseated, etc. He reduced that dosage, upped the other one! Needless to say things did not get any better. By now I was having blurred vision, light headedness, fuzzy brain, etc. so he changed the meds again. By now my blood test results were totally crazy: liver enzymes off the wall, sodium and potassium way down, BUN & creatinine out of whack. And now I was having a high pitched whine/hum in my head (not tinitus) and involuntary muscle spasms of the body, arms and legs, which would start just as I was falling asleep (needless to say, I didnt get much sleep). Due to the rapid drip in my electrolytes, I had the pleasure of spending 3 days in ICU in January and a month later, in February, they ran all sorts of blood and urine tests. The cardio doc said it was probably a problem with ADH (anti-diuretic hormone) but those were normal. He told me to restrict fluid intake to 49 oz/day and released me. The symptoms continued.
(As an aside, I was also having pins-and-needles pains in my right shoulder and arm along with cramping. Three neurologists said there was really nothing wrong, maybe some minor carpal tunnel, although one said it wasnt that, so he did an EEG and a brain MRI. Nothing wrong!)
By now I was totally frustrated. I havent worked in about three years with all these problems, but I kept hoping someone would figure this thing out! I fired my cardiologist and neurologist(s) and found an internist who listened. She ran a whole battery of tests and was still scratching her head. Fortunately, she reduced and/or changed a number of meds which reduced some of the problems, but the attacks/episodes kept on coming.
To cut a looooong story shortâ?¦.during the first week of November, 2004, I went to the Kingwood Northeast Minor Emergency Clinic for what I thought was strep throat. It turned out to be an oral yeast infection due to the antibiotics I was taking after a couple of root canals. After the yeast diagnosis the doctor asked about my skin color, kind of ruddy-reddish. He said that I didnt appear to be Native-American. I told him it had been that way for years, and I thought it was sun damage. He looked at some of my previous records and noticed some out-of-normal things with my blood tests, asked a few questions about my current symptoms, and said, I think you have acute intermittent porphyria. What? was my response. He copied some info out of a physicians reference book and suggested I have my internist run some specific tests. Sure enough, porphyria.
I called that doctor after the test results and thanked him profusely. He said I made his year! He is a certified neurologist/ophthamologist but prefers to work in urgent care.
Now I am researching everything I can about AIP. As my brother-in-law said, I will know more about porphyria than 99.9% of the doctors. I also have the resources of several doctors in my extended family, none of whom unfortunately, live anywhere near Texas, but I do have a phone/fax/e-mail.
After the diagnosis I have switched to a high complex-carb, low-moderate protein (Mediterranean) diet. Additionally, I am taking glucose tabs and as of this writing I have gone 14 days without an attack, which is the longest length of time in a long time.
Someone inboxed me today and asked the following question: What makes you so happy? What changed your life? Please shareâ?¦
Here was my reply, which I thought Id share:
I have no reason to be unhappy. Other than my health problems (admittedly of which there are many) my life is wonderful. So I choose to focus on the good. Its an active, ongoing choice, and not always an easy one. But my health issues are just a part of me, not all of me. We only get one life and Im not going to live it wallowing in sadness over the unfortunate hand Ive been dealt with my health. To me, that would be a waste of a beautiful thing. Everyone has problems, and I firmly believe that how we deal with them and how we cope are what makes the difference between happiness and unhappiness. I have bad days like anyone else, but my life is overwhelmingly good and joyful despite my health problems. So I choose to focus on those things instead.
Of course, my own body gets in my way sometimes and I do get downâ?¦ Im human. But happiness, I firmly believe, is a CHOICE. And it takes dedication and commitment just like any other important choice in life. But its so, so worth it. I think that going through something as scary as brain surgery, watching my brother lose his best friend to cancer at age 23, knowing a couple who in their early 30s are dealing with ALS, etc., have also put a lot of things into perspective for me. My faith in God and getting in touch with my spiritual side have helped, too, as well as writing/journaling and doing good for others. When I try to focus on helping other people or animals with their problems, it makes me focus less on my own.
People have said I must not be that sick if Im able to be happy, but if they are saying that, they have no idea what Ive been through. Thats almost insulting to me! But there wasnt one pivotal moment that changed my life. A lot of little things, a lot of lessons learned, books read, sermons taken to heart, quotes that have inspired, people who have changed my outlook, events for which I have been grateful and so on.
So, thats my answer in a nutshell. There was no magic pill, magic wand or magic remedy. I just made a choice to not let my health problems bring me down. Physically, I made a choice to cut back my medications. For awhile I did things 100 percent holistically and all-natural which served me better and put me into clinical remission. Now, Im thinking of slowly reintroducing just one med for maintenance purposes and to prevent dangerous complications. But other than that, my answer to your question, What makes you so happy? is this: the GIFT of LIFE. I hope this helps. And I hope you dont mind me sharing this anonymously because Im sure that others wonder as you did.
Thanks for reaching out. Best wishes and be well! Choose Hope. Choose Happy. Stay Positive.
Keeping a Positive Outlook When Dealing With Chronic Illness
Thursday - March 29, 2018 @ 11:40:47
Keeping a Positive Outlook When Dealing With Chronic Illness
How to Stay Realistic When Coping With Medical Disease
Friends and family often encourage people with illness to stay positive. Is this really helpful?
For people who are medically ill, keeping an optimistic outlook can be tough. Illness is scary and forces them to deal with uncertainty, a lack of control, surrendering to doctors, and the reality that life is finite. All of that is pretty frightening and difficult, so I am not surprised when someone cannot feel positive all of the time when coping with illness.
People who are sick do not need to feel positive all of the time.
It can be trying for people who are ill when friends and family encourage patients to feel happy or optimistic. When a well-intentioned friend says such things as, Everything happens for a reason, or I am sure that you will be stronger from this, it can (albeit, unintentionally) pressure patients into acting cheery, when they may not feel that way.
In my experience, people struggling with illness want acknowledgement about how hard their situation is. Those who are ill may not want to dwell on how difficult things are. Yet, when friends and family are overly focused on being positive, it can result in patients feeling pressured to articulate how much they are suffering! Hence the paradox: When the loved ones of people suffering with illness make space for negative emotions, hopeful feelings and better coping often follow.
Being positive all of the time when dealing with illness is unrealistic. In fact, being excessively upbeat is sometimes linked with the denial of illness. Being Pollyannish or in denial can lead to negative psychological and even physical consequences.(link is external) For example, avoiding the reality of illness can lead to people not taking care of themselves.
What I recommend is finding a balance between acting falsely buoyant and feelings of despair. Being hopeful is reasonable. Complete disavowal of negative information about illness can be problematic; cautious optimism is often ideal.
People who are ill do best when they focus on what they can controldiet, exercise, and decisions about physicians and treatments. Attitude is important as well. Being positive can be helpful; patients should just make sure there are enough loved ones who can listen when the chips are down. People who are ill need friends and family who can tolerate hearing about all kinds of feelings.
Spread the WORD! Porphyria Awareness Week will be held on April 21-28, 2018
Friday - March 23, 2018 @ 07:00:02
Porphyria Awareness Week will be held on April 21-28, 2018. We strive to dedicate this week to promote this group of rare diseases, reduce the stigma associated with porphyria through physician education, and provide support for those affected.
National Porphyria Awareness Week is the time for you to bring porphyria awareness to your local communities. We, at the American Porphyria Foundation, encourage You to help raise awareness and provide accurate information about porphyria where you live. The APF will help you accomplish your own activity by providing:
â?¢ Porphyria Brochures â?¢ Porphyria Fact Sheets â?¢ PorphyriaLive DVDs â?¢ Information to gain media attention â?¢ PowerPoint presentations, â?¢ Porphyria Awareness week Graphics for Print, Web, and Social media (See downloads below) â?¢ Press releases for local newspapers and television and Much More! Contact Edrin at the APF office today to request information to be sent to you!
Even as opioids flood American communities and fuel widespread addiction, hospitals are facing a dangerous shortage of the powerful painkillers needed by patients in acute pain, according to doctors, pharmacists and a coalition of health groups.
The shortage, though more significant in some places than others, has left many hospitals and surgical centers scrambling to find enough injectable morphine, Dilaudid and fentanyl drugs given to patients undergoing surgery, fighting cancer or suffering traumatic injuries. The shortfall, which has intensified since last summer, was triggered by manufacturing setbacks and a government effort to reduce addiction by restricting drug production.
As a result, hospital pharmacists are working long hours to find alternatives, forcing nurses to administer second-choice drugs or deliver standard drugs differently. That raises the risk of mistakes and already has led to at least a few instances in which patients received potentially harmful doses, according to the nonprofit Institute for Safe Medication Practices, which works with health care providers to promote patient safety.
In the institutes survey of hospital pharmacists last year, one provider reported that a patient received five times the appropriate amount of morphine when a smaller-dose vial was out of stock. In another case, a patient was mistakenly given too much sufentanil, which can be up to 10 times more powerful than fentanyl, the ideal medication for that situation.
In response to the shortages, doctors in states as far-flung as California, Illinois and Alabama are improvising the best they can.
Some patients are receiving less potent medications like acetaminophen or muscle relaxants as hospitals direct their scant supplies to higher-priority cases.
Other patients are languishing in pain because preferred, more powerful medications arent available, or because they have to wait for substitute oral drugs to kick in.
The American Society of Anesthesiologists confirmed that some elective surgeries, which can include gall bladder removal and hernia repair, have been postponed.
In a Feb. 27 letter to the U.S. Drug Enforcement Administration, a coalition of professional medical groups including the American Hospital Association, the American Society of Clinical Oncology and the American Society of Health-System Pharmacists said the shortages increase the risk of medical errors and are potentially life-threatening.
In addition, having diminished supply of these critical drugs, or no supply at all, can cause suboptimal pain control or sedation for patients, the group wrote.
The shortages involve prefilled syringes of these drugs, as well as small ampules and vials of liquid medication that can be added to bags of intravenous fluids.
Drug shortages are common, especially of certain injectable drugs, because few companies make them. But experts say opioid shortages carry a higher risk than other medications.
Small Mistakes Lead to Big Errors
Giving the wrong dose of morphine, for example, can lead to severe harm or fatalities, explained Mike Ganio, a medication safety expert at the American Society of Health-System Pharmacists.
Calculating dosages can be difficult and seemingly small mistakes by pharmacists, doctors or nurses can make a big difference, experts said.
Marchelle Bernell, a nurse at St. Louis University Hospital in Missouri, said it would be easy for medical mistakes to occur during a shortage. For instance, in a fast-paced environment, a nurse could forget to program an electronic pump for the appropriate dose when given a mix of intravenous fluids and medication to which she was unaccustomed.
The system has been set up safely for the drugs and the care processes that we ordinarily use, said Dr. Beverly Philip, a Harvard University professor of anesthesiology who practices at Brigham and Womens Hospital in Boston. You change those drugs, and you change those care processes, and the safety that we had built in is just not there anymore.
Chicago-based Marti Smith, a nurse and spokeswoman for the National Nurses United union, offered an example.
If your drug comes in a prefilled syringe and at 1 milligram, and you need to give 1 milligram, its easy, she said. But if you have to pull it out of a 25-milligram vial, you know, its not that were not smart enough to figure it out, it just adds another layer of possible error.
During the last major opioid shortage in 2010, two patients died from overdoses when a more powerful opioid was mistakenly prescribed, according to the institute. Other patients had to be revived after receiving inaccurate doses.
DEA must balance the production of what is needed for legitimate use against the production of an excessive amount of these potentially harmful substances, the agencysaid in August. When the coalition of health groups penned its letter to the DEA last month, it asked the agency to loosen the restrictions for liquid opioids to ease the strain on hospitals.
The shortages are not being felt evenly across all hospitals. Dr. Melissa Dillmon, medical oncologist at the Harbin Clinic in Rome, Ga., said that by shopping around for other suppliers and using pill forms of the painkillers, her cancer patients are getting the pain relief they need.
Dr. Shalini Shah, the head of pain medicine at the University of California-Irvine health system, pulled together a team of 20 people in January to figure out how to meet patients needs. The group meets for an hour twice a week.
The group has established workarounds, such as giving tablet forms of the opioids to patients who can swallow, using local anesthetics like nerve blocks and substituting opiates with acetaminophen, ketamine and muscle relaxants.
We essentially have to ration to patients that are most vulnerable, Shah said.
Two other California hospital systems, Kaiser Permanente and Dignity Health in Sacramento, confirmed theyre experiencing shortages, and that staff are being judicious with their supplies and using alternative medications when necessary.
At Helen Keller Hospitals emergency department in Sheffield, Ala., earlier this month, a 20-year-old showed up with second-degree burns. Dr. Hamad Husainy said he didnt have what he needed to keep her out of pain.
Sometime in January, the hospital ran out of Dilaudid, a drug seven times more potent than morphine, and has been low on other injectable opioids, he said.
Because Husainys patient was a former opioid user, she had a higher tolerance to the drugs. She needed something strong like Dilaudid to keep her out of pain during a two-hour ride to a burn center, he said.
It really posed a problem, said Husainy, who was certain she was in pain even after giving her several doses of the less potent morphine. We did what we could, the best that we could, he said.
Bernell, the St. Louis nurse, said some trauma patients have had to wait 30 minutes before getting pain relief because of the shortages.
Thats too long, said Bernell, a former intensive care nurse who now works in radiology.
Dr. Howie Mell, an emergency physician in Chicago, said his large hospital system, which he declined to name, hasnt had Dilaudid since January. Morphine is being set aside for patients who need surgery, he said, and the facility has about a weeks supply of fentanyl.
Mell, who is also a spokesman for the American College of Emergency Physicians, said some emergency departments are considering using nitrous oxide, or laughing gas, to manage patient pain, he said.
When Mell first heard about the shortage six months ago, he thought a nationwide scarcity of the widely used drugs would force policymakers to come up with a solution before it became dire.
But they didnt, he said.
Kaiser Health News is a nonprofit news service covering health issues. It is an editorially independent program of the Kaiser Family Foundation, which is not affiliated with Kaiser Permanente.
NEWS! CLINUVEL HOLDS EUROPEAN ERYTHROPOIETIC PROTOPORPHYRIA (EPP) EXPERT MEETING
Sunday - March 18, 2018 @ 07:00:37
CLINUVEL HOLDS EUROPEAN ERYTHROPOIETIC PROTOPORPHYRIA (EPP) EXPERT MEETING
� Expert porphyria physicians and medical staff from 12 European countries were represented � First PASS1 analyses: 13 months of data from adult EPP patients receiving SCENESSE collected through the European EPP Disease Registry � Unchanged safety profile of SCENESSE as per SmPC² � Clinical effectiveness: continuation on treatment by 99% of patients � 61% of patients were treatment naïve (no previous exposure to afamelanotide) � Over 16% of patients requested start of treatment in autumn and winter months � Positive evaluation by physicians and patients on therapeutic benefit � Requests for an afamelanotide formulation for the paediatric EPP population
Melbourne, Australia and Leatherhead, UK, 19 March 2018
CLINUVEL PHARMACEUTICALS LTD (ASX: CUV; XETRA-DAX: UR9; NASDAQ INTERNATIONAL DESIGNATION: CLVLY) today announced the results of the third European meeting on erythropoietic protoporphyria (EPP) held in Vienna, Austria, on Friday 16 March.
Physicians and medical staff from 12 countries and 21 European EPP expert centres discussed the ongoing treatment of adult patients diagnosed with EPP, the post-authorisation obligation for collection of pseudonymised data (patients identifying details are coded), and the clinical relevance of the treatment with SCENESSE (afamelanotide 16mg).
EUROPEAN POST-AUTHORISATION SAFETY STUDY (PASS) CLINUVEL is required to conduct a post-authorisation safety study (PASS), as agreed with the European Medicines Agency (EMA) in October 2014, capturing data on the ongoing safety and use of SCENESSEin adult EPP patients participating in the European EPP Disease Registry (EEDR).
Initial formal analyses of data from the EEDR consisted of approximately 34,000 patient-days- exposure to treatment between 22 June 2016 and 30 June 2017. The data showed an unchanged safety profile for SCENESSE compared to the Summary of Product Characteristics (SmPC).
As one of the measures of clinical effectiveness it was found that, as of 30 June 2017, 99% of the EPP patients were still continuing treatment. Further analyses of clinical effectiveness are being conducted for those EPP patients who continue treatment and those who received their first treatment after 30 June 2017.
Of interest was that 61% of the patients receiving SCENESSE had been treatment naïve prior to participating in the PASS, in that they had not received afamelanotide during CLINUVELs clinical trial program from 2005 to 2013, or through compassionate use or special access schemes.
Following the initial data presented during the European expert meeting discussions were held on the overall clinical experience with the medicinal drug. The general observations from the attending medical staff was that SCENESSE provided therapeutic value under real-world conditions and that it enabled EPP patients to engage in daily activities which had not been previously possible. None of the attending porphyrinologists declared a conflict of interest and all provided an opinion in an independent capacity.
Of particular interest was that under conditions of use, 16% of the EPP patients sought treatment during the autumn and winter months. Medical staff from the expert centres expressed that a higher number of parents and carers had requested afamelanotide treatment for children diagnosed with EPP since the treatment for adults had been made available. In the coming year CLINUVEL intends to discuss with the EMA suggested changes to the SmPC, in particular to widen the recommended maximum dosing of SCENESSE per calendar year.
COMMENTARY The feedback on real-life clinical experiences from EPP patients and physicians is very important to us and provides significant information for future development, CLINUVELs Director of Clinical Affairs, Dr Emilie Rodenburger said. The efforts of physicians and medical staff to treat patients under the strictly imposed post-authorisation measures need to be acknowledged. It is highly motivating and satisfying to learn first-hand the impact of the work done by CLINUVELs team in assisting patients who had not been clinically attended before. I also take away from this meeting how much experts have appreciated our teams ability to persevere under all circumstances.
- End
1 SCENESSE (afamelanotide 16mg) is approved in Europe as an orphan medicinal product for the prevention of phototoxicity in adult patients with EPP. A post-authorisation safety study (PASS) has been implemented to capture data on the ongoing use of SCENESSE in European EPP expert centres, with other measures agreed under a Risk Management Plan for SCENESSE. Information on the product can be found on CLINUVELs website at www.clinuvel.com.
2 The SCENESSE Summary of Product Characteristics (SmPC) is available on CLINUVELs website.
About CLINUVEL PHARMACEUTICALS LIMITED CLINUVEL PHARMACEUTICALS LTD (ASX: CUV; NASDAQ INTERNATIONAL DESIGNATION ADR: CLVLY; XETRA-DAX: UR9) is a global biopharmaceutical company focused on developing and delivering treatments for patients with a range of severe genetic and skin disorders. As pioneers in understanding the interaction of light and human biology, CLINUVELs research and development has led to innovative treatments for patient populations with a clinical need for photoprotection and repigmentation. These patient groups range in size from 5,000 to 45 million worldwide. CLINUVELs lead product, SCENESSE (afamelanotide 16mg), was approved by the European Commission in 2014 for the prevention of phototoxicity (anaphylactoid reactions and burns) in adult patients with erythropoietic protoporphyria (EPP). More information on EPP can be found at http://www.epp.care. Headquartered in Melbourne, Australia, CLINUVEL has operations in Europe, Switzerland, the US and Singapore. For more information go to http://www.clinuvel.com.
SCENESSE is a registered trademark of CLINUVEL PHARMACEUTICALS LTD.
Forward-Looking Statements This release to the Australian Securities Exchange and to press may contain forward-looking statements, including statements regarding future results, performance or achievements. These statements involve known and unknown risks, uncertainties and other factors which may cause CLINUVELs actual results, performance or achievements to be materially different from any future results, performances or achievements expressed or implied by the forward-looking statements. These statements reflect our current views with respect to future events and are based on assumptions and subject to risks and uncertainties. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Some of the factors that could affect the forward-looking statements contained herein include: that the FDA may require additional studies beyond the studies planned for product candidates or may not provide regulatory clearances, including for SCENESSE; that the FDA may not provide regulatory approval for any use of SCENESSE or that the approval may be limited; that CLINUVEL may never file an NDA for SCENESSE regulatory approval in the US; that the Company may not be able to access adequate capital to advance its vitiligo programs; that the Company may not be able to retain its current pharmaceutical and biotechnology key personnel and knowhow for further development of its product candidates or may not reach favourable agreements with potential pricing and reimbursement agencies in Europe and the US. Level 6, 15 Queen Street T +61 3 9660 4900 www.clinuvel.com Melbourne, Victoria 3000 F +61 3 9660 4999 Australia
CLINUVEL PHARMACEUTICALS LTD today announced the results of the third European meeting on erythropoietic protoporphyria (EPP) held in Vienna, Austria, on Friday 16 March.
EPP ~ Video Link From Clinuvel & Patient Member Story: Lauren Fabean
Thursday - March 15, 2018 @ 06:00:33
EPP Video: https://vimeo.com/17178430
Lauren Fabean
Type of Porphyria:
Erythropoietic Protoporphyria (EPP)
My name is Lauren Fabean, and I am a sun-hater! I enjoy rainy days and feel an immediate sense of relief when I see "considerable cloudiness" in a weather forecast.
I was diagnosed with EPP at the age of 12. After enduring the worst reaction of my life while participating in a weekend softball tournament (I really hated playing softball, too!), my mom started seeking answers to what was happening to me and why, and the journey to my diagnosis began. It took me a long time to get used to having EPP, and to learn what preventative measures worked for me. I also had to learn how much sunlight I could handle before a reaction set in. This was really hard. 23 years later, I still have days when I think I will be ok in a bit of sunlight, and the next thing I know I'm kicking myself for allowing a reaction to occur.
This is what happens to me when I am exposed to too much sunlight for too long of a time period: I feel an intense burning sensation in my hands, feet, and/or face. When trying to cool the areas, I feel an extreme tingling sensation if I am applying something that is too cold. I need to find a happy medium. My skin is also very sensitive during this time; a simple bump or scrape against an object leaves a lasting, uncomfortable tingling sensation. A bad reaction can take days to go away. In the summer, even if I'm very careful, I have come to terms with the fact that I will always be living with a slight reaction here or there. Unless I want to just hide inside every day.
What works for me: I am a firm believer in sunscreen, not just because of EPP, but because the sun is so damaging to our skin and it's so important to take care of it. I have spent a lot of money on good clothing with SPF and hats, and I can be seen carrying an umbrella (my "sunbrella" as I call it) at times. The best thing for me to do is simply stay away from the rays.
I struggled a bit growing up, and did feel lonely at times. However, the worst part of having EPP in my adult life is the fact that I cannot play or swim with my daughter in the summer. I get to sit on the sidelines, hiding in my shade, while I watch my husband enjoy all of those things that I wish I could do. This disease has taught me a lot. Most importantly, I realize that I am very lucky. There are many others who suffer far worse than I do when exposed to sunlight. EPP is mostly a huge inconvenience in my life, but its something that Ive accepted. So, if you see me running desperately to the shade or hiding under my hats and umbrella, dont feel bad for me or think Im weird. Thats just me living with my sun thing.
Back in late 2012, I kept getting these random, tiny blisters. When I finally said something to my primary care doctor, she looked at me and told me to quit smoking and guaranteed that the blisters will go away if I did. I should mention that the day I started going to her, she hounded me about smoking. I didn't quit smoking, but I did cut back. A few months later, I got mad at her, so she sent me to a dermatologist. I had a blister (luckily) on my finger at the time of visit. He dug it out for a biopsy and suspected I had Porphyria Cutanea Tarda (PCT). I went back 2 weeks later, and he said it was confirmed that I did have PCT. He then advised me that I was to avoid direct sunlight at all cost, told me to get some Neutrogena sunblock SPF 100+ for sensitive skin, and to invest in long sleeve t-shirts, gloves, hats, and scarves. He advised me to use the sunblock along with the "winter clothing" while I was living in Texas. With the weather being 100° or higher on some days, it seemed to be difficult. I tried as much as I could. The blisters kept coming and increasing in size. I didn't have any health insurance at the time.
My PCP was at the HIV services in Fort Worth. Since I was HIV+, that's where I went for HIV services. The dermatologist had told me I had very high iron, too much in my blood, and that a scheduled phlebotomy twice a month for 6-8 months would be the cure. Seems easy enough, right? He said he didn't do the phlebotomies and had gotten ahold of my PCP and she had told him that if they could get the supplies, then I could do the blood draws there, where I went for HIV services. In the meantime I quickly added private insurance coverage at the job I had. I was the 2nd shift lead caregiver/med passer/supervisor at an Alzheimer's and Dementia Assisted Living Community. A week or so later, my PCP told me they couldn't get the supplies for my phlebotomies, and I'd have to get a referral through the dermatologist. I did that, but insurance didn't cover it, which left me having to pay $250 twice a month for phlebotomies. There was absolutely no way I could have afforded that, and neither the PCP nor dermatologist put forth any effort to try and help me out or find other options. So, that left me to simply deal with it. Some time went by and things in my personal life changed, which resulted in me moving back to my home state of Michigan.
I was nervous of the HIV services here in Michigan (Lansing), because I rarely heard good things about them. However, they have been nothing but amazing. My PCP here quickly referred me to an oncologist at Sparrow Cancer Center in Lansing, MI. He instantly diagnosed me with Erythrocythemia, which is a more specific term for saying I have high red blood cell levels, and I told the oncologist that I was diagnosed with PCT in Texas. He told me that he could not diagnose me with that without first running tests or seeing a proof of diagnosis. His place was to do monthly phlebotomies, and I had told him that in Texas it was supposed to be twice a month. He said to start them monthly, for now. Meanwhile, my PCP wanted to diagnose me with Polycythemia. According to the doctors, the treatment, which would be phlebotomies, is the same for all three conditions. So I started my first phlebotomy back in June. I still wasn't happy that it was only once a month, so I demanded twice a month and the doctor agreed. So after 3 blood draws at going twice a month, my hemoglobin and red blood cell levels were HIGHER than when I had them tested in May, before the phlebotomies started. So starting tomorrow, August 1, 2015, I start phlebotomies twice a week, and they draw 450cc's, which is the amount they've been drawing all along. I should also add that I do have Hepatitis C that I acquired through a blood transfusion when I was born in 1979. I'm still a smoker and will be done by the end of the day today. Thanks for reading my short story.
~ John Gladding, PCT
What I Wish I Knew
Thursday - March 8, 2018 @ 07:00:41
What I Wish I Knew
When I was first diagnosed with Acute Intermittent Porphyria I was initially shocked.I struggled with the news and then I pulled myself together.I experienced many different emotions and often felt I would somehow make sense of it and get past it as soon as possible.I wanted to power through it and get my life back again.I closed myself off to lots of support along the way believing I shouldnt ask for help or expose my weaknesses.When I look back on it know, I realize though I am coping the best way I know how, there were so many things I wish I had known that would have made it easier for me, and probably my family and friends to.
Knowledge is Power: What I wish I Knew
Im not going to talk to anyone about my Porphyria, I said
Have you ever said that?
Where can I Find Information
I felt lost, scared, emotional, could my family really understand?
Did they want to understand?
Did they believe me?Did my Doctor even believe me?
Now that I understand what care is needed I asked myself?
Who am I going to find to Bring with ME? Do they want to go?Will they support me.Will they say I am crazy?
I have Never asked for Help Before!
Its too hard trying to communicate with my family, friends with phone calls and texts while Im so sick, why bother I asked myself?
Do I need a special group to go to?They will think Im nuts��
No Im just going to stay inside and sleep it off.
Is this stress, or anxiety, fear, am I going to die?
What am I going to do? Who will I turn to who will help me?
I often ask myself these things and its important to journal, talk to a trust friend and family member, pray, start searching and dont stop until you get an answer good or bad, learn to be patient and kind.Be firm when you should, be your own advocate! Teach, read, watch do all that you can so you can conquer correct your lifestyle do what you must do, so that at the end of the day you can say I have done my very best!Dont stop there.Start all over when you wake upâ?¦..
Amy L Chapman
Acute Intermittent Porphyria
I will survive!
Panhematin Patient Assistance Program
Tuesday - March 6, 2018 @ 14:13:26
Panhematin Patient Assistance Program
IMPORTANT NOTICE!
If you have had problems receiving Panhematin treatment for any reason, please contact Desiree Lyon Howe, APF Executive Director, at lyonapf@aol.com .
Please note that there is a patient assistance program that was developed to help patients who experience issues with access to treatment and offers comprehensive assistance for insurance related issues.
Contact 1.866.209.7604 to access this program.
Below are a few problems that have been brought to our attention. We hope to help.
Some people have been admitted to the hospital expecting treatment and received none.
Some have been given only one vial instead of the usual 4.
Some have been told the treatment is too expensive to order or that it is only for very serious cases.
Some have standing orders for Panhematin and the orders were ignored.
Others have heard that Panhematin will take too long to arrive, when it can arrive within 16 hours of ordering.
Others have been told in the ER that there is nothing that can be done for their disease.
The excuses to treat are plentiful. Please note that every attack can be serious and should be treated per the doctor.
Please let us know your situation.
Upcoming Events
The APF Office will be closed on Monday, February 19, 2018 and will reopen for normal business hours on Tuesday, February 20, 2018.
Research
Givosiran Trials
The phase 3 trials for Givosarin have started. The purpose of this study is to evaluate the use of this medication to prevent or reduce attacks and symptoms in those with an acute porphyria (AIP, HCP, VP, ADP). If you are interested in participating for this exciting new trial, call Edrin at the APF 713.266.9617 or 1.866.APF.3635
Hello and welcome to the American Porphyria Foundation. Many of you are NEW to the APF! Welcome!!! We are so happy that you are here to learn about Porphyria disorder and how really rare YOU are. Our information and links provided are backed by Worldwide Porphyria Experts! There are many modern day sites and theories about Porphyria and social media groups that may offer personal help or experience. Caution should be used when looking at information and considering the source. Ask yourself, Who gave this information to me? Where did I read such? Is this a patient themselves, an advocate or a medical professional? The more Research and time you put into knowing more about your disease you will benefit and learn the most! As you read and talk to different ones in the medical community be prepared by writing down in advance your questions? Why are you asking this question? Do I personally experience this problem? Talk to your Dr! Be brief and to the point! If your physician is not aware about Porphyria please call our office we would be most happy to send a free comprehensive Dr Kit to them. Our number is 1/866/APF/3635. Here are a few links to help you be more informed: http://www.porphyriafoundation.com/about-porphyria/types-of-porphyria/AIP http://www.porphyriafoundation.com/about-porphyria/types-of-porphyria/VP http://www.porphyriafoundation.com/about-porphyria/types-of-porphyria/HCP http://www.porphyriafoundation.com/about-porphyria/types-of-porphyria/ADP http://www.porphyriafoundation.com/about-porphyria/types-of-porphyria/PCT http://www.porphyriafoundation.com/about-porphyria/types-of-porphyria/EPP-and-XLP http://www.porphyriafoundation.com/about-porphyria/types-of-porphyria/CEP http://www.porphyriafoundation.com/about-porphyria/types-of-porphyria/HEP
Basic Genetics
To find out more information on the basic genetics of Porphyria visit:
My name is Gregary Edwards, when I was 15 is when I noticed my first issues one morning I just could not get going, I thought I just had a bug and it would go away then I started to get electric shocks shooting all throughout my body in my muscles and my bones. I was getting so weak that I had to quit football as I could no longer keep up through the pain and weakness. I had good days and bad but I could never get back to what I was as a football player. After moving schools across country I felt better enough to go out for football again but that did not last. Things were fine for awhile but during my junior year things acted up again. The shocking came back so bad it would scare other kids in class with my jumping and jerking. It would bring tears to my eyes. One day during Trig class I zoned out and the teacher sent me to the office but I collapsed in the doorway for the first time ever. And for the first time ever I was taken to the ER. Where of course everything was normal and being just poor kid no doctor wanted to pursue anything else. I noticed that when I was the weakest I had started to have more and more abdominal pain and I would swing back and forth between severe constipation and diarrhea. My weakness was getting so weird; I seemed to be strong but my endurance was poor where it used to be high. I could start off doing anything with strength but soon I would end up shaking and trembling so bad I could no longer use the muscle. If I tried to push through it like they always say "no pain no gain", my muscles would give out completely and I potentially would loose control of them. It was like they weren't even connected to my body. Over the next ten years I would go through these bouts of getting completely out of it and back to fine again but the longer it went on the less healing I would do and would remain weak and in pain.
After I graduated college and started teaching things were better; I was less stressed but as time went on and I had to go back to school while still teaching I was only getting a few hours of sleep at night and not eating much, my symptoms came back with a vengeance. I had small fiber peripheral neuropathy diagnosed. I ended up using forearm crutches and canes to help me walk. I had MRIs of my brain that showed these holes that would come and go in size and shape and one neurologist would say it's the cause of my migraines and another said it was the beginning of a demyelinating disorder. I started having seizures. My leg weakness was so constant I started to use an electric wheelchair when I was not in tight spaces where I could have help to hold myself up. I eventually moved again to be a college professor of anatomy. I was out of the wheelchair for now and felt excited to be doing what I wanted to be doing, however I still had to finish my doctoral thesis as a result I started to ignore my health. Everything was making me nauseous and cause immediate vomiting. My abdomen was in constant pain and I ended up back in my wheelchair and was having seizures. They occurred during class so I was let go from the college. I am on federal disability now still in pain, still having seizures (as what happened yesterday when I collapsed in the doctors office had three seizures and collapsed again in the ER), still getting weak and damaging my muscles, still sick from eating certain foods I find new ones all the time. I did find out what has been the cause of my migraines and seizures though dehydration and electrolyte I'm balances both happen during an attack and both are what leads to brain damage, if it is severe enough it will cause the blood brain barrier to shrink tight in certain areas leading to the symptoms and the longer it persists the more permanent damage is caused.
Three years ago was when the term porphyria was first mentioned to me as a possible answer to my years of suffering. I always felt sick so it was hard to get the samples during the worst part of an attack, sometimes you just never knew, so it has taken multiple tests. When I first got a good result my doctor told me it seemed likely to be porphyria, but with many kinds of acute porphyria more testing was needed. At first the testing indicated probable Acute Intermittent Porphyria or AIP however further testing has just confirmed it to be Hereditary Coproporphyria or HCP. Now I need to have all my family tested, with 6 kids of varying symptoms starting, then two brothers, and my mother. Education is the best defense against the ignorance that leads to 25 years of suffering.
Rare Disease Day takes place on the last day of February each year. The main objective of Rare Disease Day is to raise awareness amongst the general public and decision-makers about rare diseases and their impact on patients' lives.The campaign targets primarily the general public
and also seeks to raise awareness amongst policy makers, public authorities, industry representatives, researchers, health professionals and anyone who has a genuine interest in rare diseases.
Why?
Building awareness of rare diseases is so important because 1 in 20 people will live with a rare disease at some point in their life. Despite this, there is no cure for the majority of rare diseases and many go undiagnosed. Rare Disease Day improves knowledge amongst the general public of rare diseases while encouraging researchers and decision makers to address the needs of those living with rare diseases.
WHO?
Rare Disease Day events are down to hundreds of patient organisations all over the world who work on a local and national level to raise awareness for the rare disease community in their countries.
Since Rare Disease Day was first launched by EURORDIS and its Council of National Alliances in 2008, thousands of events have taken place throughout the world reaching hundreds of thousands of people and resulting in a great deal of media coverage. We especially thank our official Rare Disease Day partners, the National Alliances. These are umbrella organisations who group together several rare disease organisations in a given country or region. Click on a logo of one of the National Alliances to go to their website.
WHERE?
The campaign started as a European event and has progressively become a world phenomenon, with the USA joining in 2009 and participation in 94 countries all over the world in 2017. Hundreds of cities continue to take part in Rare Disease Day and we hope even more will join in 2018.
Some countries have decided to raise rare disease awareness further, for example, Spain declared 2013 as the National Year for Rare Diseases.
WHEN?
The first Rare Disease Day was celebrated in 2008 on 29 February, a rare date that happens only once every four years. Ever since then, Rare Disease Day has taken place on the last day of February, a month known for having a rare number of days.
On rarediseaseday.org you can find information about the thousands of events happening around the world on the last day of February. If you are planning an event, register your event details on the Post your Event page to get your event listed on the site!
Porphyria & APF
Wednesday - February 21, 2018 @ 11:08:04
Is porphyria genetic?
Porphyria is a genetic disorder that is caused by an abnormality in the heme production process, which results in an accumulation of heme precursors.
Is porphyria hereditary?
acute intermittent porphyria (AIP) a hereditary, autosomal dominant, form of hepatic porphyria manifested by recurrent attacks of abdominal pain, gastrointestinal dysfunction, and neurologic disturbances, and by excessive amounts of δ-aminolevulinic acid and porphobilinogen in the urine; it is due to an abnormality of pyrrole metabolism.
Which doctors treat porphyria?
Because Porphyria is so rare, few physicians have experience treating patients with the disease. Most patients are in fact treated But the APF can help by putting your doctor's office in touch with a Porphyria specialist who can offer guidance on your care.
Please give us a call today if you need a comprehensive Physician Packet. Would you like to know the facts about the types?
Do you need to find a physician or an expert near you. When you contact the APF ask for Edrin.
Do you want to know how to get the free DNA? Call APF!
Does your physician have questions? Call the APF we can put them in touch with an Expert Porphyria Specialist!!
Want to know how to get involved with Research give us call!!!
ITS ALL FREE. What are you waiting for? 1/866/APF/3635
Bright Ideas for Shadow Jumpers EPP
Friday - February 2, 2018 @ 06:00:39
Bright Ideas!
This is a place for kids to learn from others with EPP, to share their own tips and tricks on things like what to wear, what to do for fun, insight for parents and so much more. We want to add YOUR tips and tricks on managing EPP. If you are a caregiver, please ask your child with EPP to share their ideas through you. These could include
·What clothes and trends do you use when covering up from the sun?
·What are the ways you adapt to do certain activities outside?
·What are fun things you do to pass the time inside while the sun is out?
·What have been your best vacations?
·How do you manage in the car? Learning to drive?
·What do you do when you feel sad or left out?
·How do you manage going to school?
·How do you describe your EPP to other people?
·and anything else you think may be helpful to others!
·I wear an oversized hoodie the hood covers the sides of my face. (Brady, age 12)
·Cut thumbholes in your sleeves to keep your jacket or sweatshirt over your hands without sliding up. This works great for young kids! (anonymous)
·Wear a big hat and walk on the shady side of the street! (Brenda, EPP)
·I have a bag prepared with a long sleeved shirt or light jacket, hat, gloves, and face cover (I use a bandanna, some use a buff) that I take with me on cloudy days so If it clears up I'm covered and don't put myself in DANGER. (Rob, EPP)
·Our son wears a zip-up lightweight jacket with a hood in the summer so that more he can unzip it to get more air and stay cooler.
·Buy inexpensive sports gloves (for example, at Five and Below). We cut the tips off the fingers so that he can still manage things with his hands.
QUICK SHADE!:
·Use a taller person as shade (Rob, EPP)
·Walk next to buildings for shade (anonymous)
·Tell teachers, coaches, other parents, etc. that THEY are your childs quickest protection�just stand between them and the sun! (Kristen, caregiver)
·Hold their hands to protect them from the sun especially when you dont have any gloves.
·Always put your back to the sun.
SCHOOL:
·Meet with teachers at the beginning of each year to explain EPP and offer guidelines for safety (caregiver)
·Initiate a 504 plan for your child. (Ask the APF for a copy of a sample plan!)
·Make sure your teachers know that your child needs a safe place for fire drills, recess, physical education class and field trips.
DRIVING:
·Carry a blanket or towel in your car at all times and close it in the top of the window to provide shade. (Kristen, caregiver)
·Have your car windows tinted with as dark as possible tinting that is allowed in your area.
VACATIONS:
·We always bring along a canopy on vacation. We can set it up wherever our son is to provide him some safe shade. (EPP caregiver)
THINGS TO DO INSIDE:
·So often during the summer I would go to the movies while all my friends were away during the day at camp. From the popcorn to the cool temperature to combat the hot summer days, I would spend hours at a time throughout the week at my local theater.The downside? So many movies meant so many tickets which meant so much money spent.Thats why, with this new thing called MOVIE PASS, going to the movies has been more affordable. For one flat monthly fee of $9.95, movie passer customers can see one movie a day each day for the month. To sign up and too see theaters in your area qualify go to https://www.moviepass.com/
THINGS TO AVOID:
·Remember that water and snow reflect the sun!
OTHER:
·Hey Shadow Jumpers! Heres a cool tip I wanted to pass along� (Craig, EPP)
People with EPP often check the UV index every day. I know I regularly check the UV index for the day to know what I should wear or think about doing. So often people traditionally check the newspaper or kids rely on their parents. Today I wanted to share with you some great UV Apps!
Wolfram Sun Exposure Reference App Based on your skin type, what SPF youre wearing and the UV forecast for your location, this comprehensive app can predict exactly how long you can stay in the sun before burning. Organizing a trip to the shore? Check out a five-day UV forecast displayed on a map, and find out what hours each day you should minimize sun exposure. ($0.99; available for iOS) Ultraviolet ~ UV Index Keep things simple with this cool tracker, which displays the current UV index in your area using a large, vibrantly colored circle. Blues and greens mean youre in the clear while reds and purples mean a dangerously high index. General sun safety advice will tip you off for when its time to put on a hat, apply sunscreen or avoid going outside altogether. Just the bare necessities for when youre bare at the beach. (Free; available for iOS)
EPAs SunWise UV Index The U.S. Environmental Protection Agency designed this easy-to-navigate application, which delivers location-based UV index information. Ideal for plan-ahead types, the most useful feature is the color-coded hourly forecast that makes it easy to spot when the UV index is highest. (Free; available for iOS and Android)
(PLEASE NOTE: This is a good tip to check the weather, but EPP reactions occur from visible light. UV Index applications and apps will ONLY indicate ultra-violet light, not the visible light range that affects EPP)
Introducing Lighting the Moment 2018
Wednesday - January 31, 2018 @ 23:26:13
Introducing Lighting the Moment 2018
Introducing Lighting the Moment 2018
Some of the best moments we can have in our lives tend to come on vacation. We go to places we otherwise would have never gone to, we learn about cultures and traditions that once seemed so foreign and we grow through experiences that later become summed up by in inside jokes, gotta be there moments and with the people we took those trips with. And what is more memorable than those classic family trips with the family? The ones that live on in old photos, admired for the absurd fashion choice of the decade and those classic tales of who got sick at the worst time, who was unamused by the Grand Canyon, and so on. It is our belief that a vacation, more so a family vacation.
Familys come in all forms, but every family deserves the same chance at doing and experiencing a great family vacation. And every family deserves to do that regardless of the sun. Thats why we at Shadow Jumpers have introduced Light the Moment 2018, a chance to help families with EPP go out and do something for a vacation they otherwise wouldnt have tried. Let us do our best to show you the sun is worth taking on, if you know the right tricks, strategy and creativity to do it.
This year we will be sending our first family on a vacation this coming summer to the most magical place on earth, Disney World in Orlando Florida. The whole immediate family will be flown all expenses paid and put up in one of Mickeys and Minnies favorite resorts. From there, be treated to access to every park. From the awe of Cinderellas castle, to looking for captain Jack Sparrow or from blasting off with Buzz Lightyear and diving down with Nemo, there is something for everyone during the whole trip. With our help on food, merchandise and some surprises along the way, let us show you every family vacation, regardless of sun, is worth trying.
We will be sun-proofing this family every step along the way. From transportation while traveling in the park and cutting all lines in the sun to extended late hours in the park with the sun down, while also providing any & all clothing, let us worry about how you get from A to B while you worry about which ride to go on for the second and third times.
Check out the application here below. We want to hear from you and your family. Tell us about you, what makes your family that special kind of family? BE CREATIVE, show us what you got! Send photos, videos, songs, for goodness sake GET MAGICAL! The application deadline is MARCH 23rd.
Starting 1/31/18-2/1/18 Midnight. Purchase any of the following Long Sleeve Tees, Or PULLOVER Hoodies, Hats, wristbands receive one free. Please NOTE: Sizes & Price Change for XL, 2XL, 3XL. This EXCLUDES THE ZIP UP HOODIES IN GRAY OR PURPLE. & Magnets-
International Orders- to UK, Canada, Europe and AUS additional Postage depending on Country by weight
Show that you are a Porphyria Warrior!!
Porphyria Cutanea Tarda (PCT) What it is and Isn't
Monday - January 29, 2018 @ 06:00:29
Porphyria Cutanea Tarda (PCT)
This disease is the most common of the porphyrias and results from a deficiency of the enzyme uroporphyrinogen decarboxylase (UROD). PCT is essentially an acquired disease, but some individuals have a genetic (autosomal dominant) deficiency of UROD that contributes to development of PCT. These individuals are referred to as having "familial PCT". Most individuals with the inherited enzyme deficiency remain latent and never have symptoms.
PCT is one of the hepatic porphyrias. Large amounts of porphyrins build up in the liver when the disease is becoming active. The disease becomes active when acquired factors, such as iron, alcohol, Hepatitis C Virus (HCV), HIV, estrogens (used, for example, in oral contraceptives and prostate cancer treatment) and possibly smoking, combine to cause a deficiency of UROD in the liver. Hemochromatosis, an iron overload disorder, also can predispose individuals to PCT.
Symptoms
The symptoms of PCT are confined mostly to the skin. Blisters develop on sun-exposed areas of the skin, such as the hands and face. The skin in these areas may blister or peel after minor trauma. Increased hair growth, as well as darkening and thickening of the skin, may also occur. Neurological and abdominal symptoms are not characteristic of PCT.
Liver function abnormalities are common but are usually mild, although they sometimes progress to cirrhosis and even liver cancer. PCT is often associated with Hepatitis C infection, which can also cause these liver complications. However, liver tests are generally abnormal even in PCT patients without Hepatitis C infection.
Diagnosis
The preferred screening test for PCT is a measurement of porphyrins in plasma. This can differentiate PCT from Variegate Porphyria. The patterns of porphyrins in urine (predominately uroporphyrin and 7-carboxylate porphyrin) and feces (predominately isocoproporphyrin) help to confirm the diagnosis. The presence of an inherited deficiency of UROD can be demonstrated by measuring the enzyme in red blood cells and is present in about 20% of patients with PCT.
Treatment and Prognosis
PCT is the most treatable of the porphyrias. Treatment seems to be equally effective in familial and non-familial PCT. Factors that tend to activate the disease should be removed. The most widely recommended treatment is a schedule of repeated phlebotomies (removal of blood), with the aim of reducing iron in the liver. This actually reduces iron stores throughout the body. Usually, removal of only 5 to 6 pints of blood (one pint every one to two weeks) is sufficient, which indicates that iron stores are not excessively increased in most PCT patients. The best guides to response are measurements of serum ferritin and plasma porphyrins. Phlebotomies are stopped when the ferritin falls to -~20ng/ml. Another treatment approach is a regimen of low doses of either chloroquine (125mg twice weekly) or hydroxychloroquine (100mg twice weekly). Usual dosages of these drugs should not be used because they can cause transient but sometimes severe liver damage and worsening of photosensitivity in PCT patients.
After treatment for PCT, periodic measurement of plasma porphyrins may be advised, especially if a contributing factor such as estrogen exposure is resumed. If a recurrence does occur, it can be detected early and treated promptly. The treatment of PCT is almost always successful, and the prognosis is usually excellent.
PCT, Hepatitis C Virus and HIV
Because PCT is frequently associated with Hepatitis C Virus (HCV) infection, it is worth noting the issues involved in treating a patient with both PCT and HCV infection.
Infection with HCV is much more common than PCT, and most people with HCV do not have PCT. However, at least in some locations, as many as 80 percent of individuals with PCT are infected with HCV. Therefore, HCV needs to be added to the list of factors that can activate PCT alongside alcohol, iron and estrogens. Other hepatitis viruses are seldom implicated in PCT, and it is not known how HCV activates PCT.
There are several different viruses that cause hepatitis. A blood test for HCV infection has not been available for very long. HCV is most readily transmitted from one person to another by blood products. Although most people who are infected with HCV have a history of exposure to blood or needles contaminated with blood, in some cases it is not known how the infection was acquired. HCV (unlike the Hepatitis B Virus and HIV) is seldom transmitted by sexual contact. It is also not readily transmitted by casual contact with other people. Therefore, people infected with HCV are not hazardous unless they somehow expose others to their blood.
It is recommended that patients with PCT be tested for HCV infection. This is done by a blood test that detects antibodies to the virus. If HCV infection is found, it may not change the treatment of PCT (by phlebotomy or low-dose chloroquine). Treatment for PCT is highly successful even in patients with HCV. Therefore, it is reasonable to treat the PCT first and then look into treatment for HCV later.
There are reasons not to treat the HCV infection before treating the PCT. HCV treatment with alpha-interferon and ribavirin is available but is often not effective. Also, liver damage progresses slowly if at all in many people with HCV. However, once the PCT is in remission it is important to assess the amount of liver damage the virus has already caused and to have follow-up visits to a doctor to monitor the liver. In some cases it may be important to treat HCV infection to try and prevent progressive liver damage.
NORD gratefully acknowledges Ashwani K Singal, MD, MSc, Division of Gastroenterology and Hepatology, University of Alabama at Birmingham, for assistance in the preparation of this report.
Synonyms of Porphyria Cutanea Tarda
UROD deficiency
uroporphyrinogen decarboxylase deficiency
Subdivisions of Porphyria Cutanea Tarda
familial porphyria cutanea tarda (PCT type 2)
sporadic porphyria cutanea tarda (PCT type 1)
General Discussion
Summary
Porphyria cutanea tarda (PCT) is a rare disorder characterized by painful, blistering skin lesions that develop on sun-exposed skin (photosensitivity). Affected skin is fragile and may peel or blister after minor trauma. Liver abnormalities may also occur. PCT is caused by deficient levels of an enzyme known as uroporphyrinogen decarboxylase (UROD). In approximately 75% to 80% of cases this deficiency is acquired (PCT type 1 or sporadic PCT); in the remaining cases, individuals have a genetic predisposition to developing the disorder, specifically a mutation in the UROD gene (PCT type 2 or familial PCT). Most individuals with this genetic mutation do not develop PCT; the mutation is a predisposing factor and additional factors are required for the development of the disorder in these individuals. These factors are called susceptibility factors and are required for the development of both sporadic and familial PCT. Generally, PCT develops in mid to late adulthood. In extremely rare cases, individuals have mutations in both UROD genes. This autosomal recessive form of familial PCT is known as hepatoerythropoietic porphyria (HEP). HEP occurs in childhood and is usually more severe than PCT types 1 or 2. NORD has a separate report on HEP.
Introduction
PCT belongs to a group of disorders known as the porphyrias. This group of at least seven disorders is characterized by abnormally high levels of porphyrins and porphyrin precursors due to deficiency of certain enzymes essential to the creation (synthesis) of heme, a part of hemoglobin and other hemoproteins. There are eight enzymes in the pathway for making heme and at least seven major forms of porphyria. The symptoms associated with the various forms of porphyria differ. It is important to note that people who have one type of porphyria do not develop any of the other types. Porphyrias are generally classified into two groups: the hepatic and erythropoietic types. Porphyrins and porphyrin precursors and related substances originate in excess amounts predominantly from the liver in the hepatic types and mostly from the bone marrow in the erythropoietic types. Porphyrias with skin manifestations are sometimes referred to as cutaneous porphyrias. The term acute porphyria is used to describe porphyrias that can be associated with sudden attacks of pain and other neurological symptoms. Most forms of porphyria are genetic inborn errors of metabolism. PCT is an acquired liver disease, in which some individuals have a genetic predisposition to developing the disorder.
Signs & Symptoms
The symptoms of PCT can vary greatly from one individual to another. Skin abnormalities characterize this disorder. Affected individuals are abnormally susceptible to damage of the skin from sunlight (photosensitivity). Extremely fragile skin that can peel or blister on minimal impact is common. Affected individuals may develop blistering skin lesions on areas of the skin that are frequently exposed to the sun such as the hands and face. These lesions may crust over.
Eventually, scarring may develop and affected skin may darken (hyperpigmentation) or fade (hypopigmentation) in color. Abnormal, excessive hair growth (hypertrichosis), especially on the face may also occur. The hair may be very fine or coarse and can differ in color. In some patients, their hair may grow, thicken and darken. Small bumps with a distinct white head (milia) may also develop, especially on the backs of the hands.
In some cases, the skin in affected areas may thickened and harden, resembling a condition known as sclerosis, this is sometimes known as pseudosclerosis. Pseudosclerosis in individuals with PCT appears as scattered, waxy, harden patches or plaques of skin.
Liver abnormalities may develop in some affected individuals including the accumulation of iron in the liver (hepatic siderosis), the accumulation of fat in the liver (steatosis), inflammation of certain parts of the liver (portal triaditis), and thickening and scarring around the portal vein (periportal fibrosis). Affected individuals may be at a greater risk than the general population of developing scarring of the liver (cirrhosis) or liver cancer known as hepatocellular carcinoma. Advanced liver disease is uncommon, except in older individuals with recurrent disease. In some cases, liver disease is due to an associated condition such as hepatitis C infection.
Causes
PCT is a multifactorial disorder, which means that several different factors such as genetic and environmental factors occurring in combination are necessary for the development of the disorder. These factors are not necessarily the same for each individual. These factors contribute either directly or indirectly to decreased levels or ineffectiveness of an enzyme known as uroporphyrinogen decarboxylase (UROD) within the liver. When UROD levels in the liver decrease to approximately 20% of normal levels, the symptoms of PCT may develop.
The UROD enzyme is essential for breaking down (metabolizing) certain chemicals in the body known as porphyrins. Low levels of functional UROD result in the abnormal accumulation of specific porphyrins in body, especially within the blood, liver and skin. The symptoms of PCT occur because of this abnormal accumulation of porphyrins and related chemicals. For example when porphyrins accumulate in the skin, they absorb sunlight and enter an excited state (photoactivation). This abnormal activation results in the characteristic damage to the skin found in individuals with PCT. The liver removes porphyrins from the blood plasma and secretes it into the bile. When porphyrins accumulate in the liver, they can cause toxic damage to the liver.
The exact, underlying mechanisms that cause PCT are complex and varied. It is determined that iron accumulation within the liver plays a central role in the development of the disorder in most individuals. Recently, researchers have discovered that a substance called uroporphomethene, which is an oxidized form of a specific porphyrin known as uroporphyrinogen, is an inhibitor that reduces the activity of the UROD enzyme in the liver. The oxidation of uroporphyrinogen into uroporphomethene has been shown to be iron dependent, emphasizing the importance or elevated iron levels in the development of PCT.
The relationship between iron levels and PCT has long been established and PCT is classified as an iron-dependent disease. Clinical symptoms often correlate with abnormally elevated levels of iron in the liver (iron overloading). Iron overloading in the liver may only be mild or moderate. The exact relationship between iron accumulation and PCT is not fully understood, however, as there is no specific level of iron in the liver that correlates to disease in PCT (e.g. some individuals with symptomatic PCT have normal iron levels).
There is an increased prevalence of mutations in the HFE gene in individuals with PCT. Mutations in the HFE gene can cause hemochromatosis, a disorder characterized by the accumulation of iron in the body, especially the liver. Hemochromatosis occurs when a person inherited two mutated HFE genes (one from each parent). Hemochromatosis is associated with low levels of hepcidin, a specialized protein that is the primary regulator of iron absorption in the body, including regulating the uptake of iron by the gastrointestinal tract and liver.
Additional risk factors that have been associated with PCT include alcohol, certain infections such as hepatitis C or HIV, and drugs such as estrogens. Some studies have indicated that smoking is a risk factor for PCT in susceptible individuals. Less often, certain chemical exposures (e.g. hexachlorobenzene), kidney dialysis, and lupus appear to be connected to the development of PCT. It is believed that these susceptibility factors reduce hepcidin in the body and consequently lead to iron accumulation in the liver. However, the exact relationship among most susceptibility factors with the development of symptoms in PCT is not fully understood. For example, alcohol clearly contributes to the development of the disorder in some cases, but PCT is not common in alcoholics. Most individuals with PCT have three or more susceptibility factors present.
In some cases, individuals develop PCT without a known susceptibility factor, suggesting that additional, as yet unidentified risk factors exist.
The underlying cause of UROD deficiency in the acquired form of PCT is unknown. Affected individuals have approximately 50% residual UROD activity and do not develop symptoms unless additional factors are present. The most common factors associated with acquired PCT are hemochromatosis or chronic hepatitis C infection. In individuals with acquired PCT, UROD levels are only deficient in the liver.
In the familial form of PCT, individuals have a mutation in the UROD gene. This mutation is inherited as an autosomal dominant trait. Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary for the appearance of the disease. The abnormal gene can be inherited from either parent, or can be the result of a new (de novo) mutation in the affected individual with no family history. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy regardless of the sex of the resulting child.
The UROD gene creates (encodes) the UROD enzyme, which is the fifth enzyme in the heme synthesis pathway. A mutation in one of these genes leads to abnormally low levels of this enzyme in all tissues of the body (not just the liver). However, one mutation alone is insufficient to cause familial PCT as residual UROD enzyme levels remain above 20% of normal. In fact, most individuals with a mutation in the UROD gene do not develop the disorder. Additional factors must be present for the disorder to develop.
Affected Populations
PCT is a rare disorder that affects males and females. The disorder usually develops after the age of 30 and its onset in childhood is rare. PCT is found worldwide and in individuals of all races. The prevalence is estimated to be approximately 1 in 10,000 to 25,000 individuals in the general population. PCT is the most common form of porphyria.
Related Disorders
Symptoms of the following disorders can be similar to those of PCT. Comparisons may be useful for a differential diagnosis.
Variegate porphyria is a rare genetic metabolic disorder characterized by deficient function of the enzyme protoporphyrinogen oxidase (PPO or PPOX). This deficiency is caused by heterozygous mutations in the PPOX gene, and leads to the accumulation of certain chemicals called porphyrins and porphyrin precursors in the body, which, in turn, can potentially result in a variety of symptoms. Specific symptoms can vary greatly from one person to another. Some affected individuals present with skin symptoms, some with neurological symptoms and some with both. Blistering and fragility of sun-exposed skin are the most common skin (cutaneous) symptoms. Common neurological symptoms include abdominal pain, nausea, vomiting, constipation, extremity pain and weakness, anxiety, restlessness and convulsions. Many different PPOX mutations have been identified in different families with variegate porphyria. The genetic mutation in a family is inherited as an autosomal dominant trait, but many individuals who inherit a PPOX mutation do not develop any symptoms (asymptomatic). (For more information on this disorder, choose variegate porphyria as your search term in the Rare Disease Database.)
Hepatoerythropoietic porphyria (HEP) is an extremely rare genetic disorder characterized severe deficiency of the enzyme, uroporphyrinogen decarboxylase. Onset is usually during infancy or early childhood, although adult onset has been reported. Affected individuals develop painful, blistering skin lesions that develop on sun-exposed skin (photosensitivity). Cutaneous photosensitivity is usually more severe in HEP than in PCT. Affected areas of skin can scar and become discolored. There is a risk of bacterial infection. Hypertrichosis is also common. Mild anemia and enlargement of the liver and/or spleen (hepatomegaly) have also been reported. Adult onset, mild cases of HEP may be clinically indistinguishable from PCT. HEP is caused by mutations of the UROD gene and is inherited as an autosomal recessive trait. (For more information on this disorder, choose hepatoerythropoietic porphyria as your search term in the Rare Disease Database.)
Pseudoporphyria is a rare skin disease that occurs upon exposure to sunlight. Affected skin may be extremely fragile. In addition, affected individuals can develop large blisters filled with a clear fluid (bullae), small bumps with a distinct white head (milia), and scarring of affected areas. Lesions form in sun-exposed areas of the skin or at the site of trauma on the skin. A sunburn-like rash can develop in some affected individuals. The skin lesions of pseudoporphyria closely resemble those seen in cutaneous forms of porphyria including porphyria cutanea tarda. Pseudoporphyria can occur at any age. Women are affected more often than men. Pseudoporphyria is caused by the use of certain medications. The disorder can also be associated with often chronic kidney failure and hemodialysis. In some cases, tanning beds or ultraviolet light therapy (phototherapy) can worsen symptoms.
There are other conditions that may cause signs and symptoms that are similar to those seen in porphyria cutanea tarda. Such conditions include other cutaneous porphyrias, drug-induced photosensitivity, various forms of lupus, and solar urticarial. (For more information on these disorders, choose the specific disorder name as your search term in the Rare Disease Database.)
Diagnosis
A diagnosis of PCT is based upon identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation, and a variety of specialized tests.
Clinical Testing and Workup
Screening tests can help diagnosis PCT by measuring the levels of certain porphyrins in blood plasma. This test can differentiate PCT from variegate porphyria and erythropoietic protoporphyria. Screening tests can also be performed on the urine or feces. The patterns of porphyrins in urine (predominately uroporphyrin and 7-carboxylate porphyrin) and feces (predominately isocoproporphyrin) help to confirm the diagnosis. Familial PCT can be diagnosed by the presence of a reduced amount of the UROD enzyme in red blood cells (erythrocytes). Molecular genetic testing is available for familial PCT if the diagnosis has been confirmed in the patient or a family member by urinary porphyrin analysis and/or enzyme assay of UROD activity.
Standard Therapies
Treatment The treatment of PCT is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, general internists, hematologists, dermatologists, hepatologists, and other healthcare professionals may need to systematically and comprehensively plan an affect childs treatment.
PCT is the most treatable form of porphyria and treatment appears equally effective for both the sporadic and familial forms. The standard treatment of individuals with PCT is regularly scheduled phlebotomies to reduce iron and porphyrin levels in the liver. This is the preferred treatment of affected individuals at many porphyria centers regardless of whether there is confirmed iron overload. A phlebotomy is a simple and safe procedure that involves removing blood via a vein (bloodletting). Since much of the iron in the body is present in red blood cells, regular phlebotomies can reduce excess iron levels in the body. Regularly scheduled phlebotomies usually results in complete remission in most individuals. A phlebotomy schedule is recommended to achieve a target ferritin level of less than 20 nanograms per milliliter (<20 ng/mL). Ferritin is an iron compound that is used an indicator of the bodys iron stores. Most patients require between five and eight phlebotomies to achieve remission.
In some cases, affected individuals may be treated with low doses of chloroquine and hydroxychloroquine, which can also reduce iron levels in the liver. These drugs are often used to treat malaria (antimalarials). This therapy is usually reserved for individuals for whom phlebotomies are not an option (e.g. contraindicated) such as in individuals with anemia, if there is the non-availability of venous access, or because of patient choice. The dosage of these drugs is especially important; dosages approaching those commonly used to treat individuals with other conditions can cause significant adverse effects in individuals with PCT including elevating porphyrin levels and worsening photosensitivity. The recommended dosages are 100 mg twice a week for hydroxychloroquine or 125 mg twice a week for chloroquine. Such a low dose schedule is equally effective as phlebotomy and easier to take with less treatment cost involved. The mechanism of action of these drugs in individuals with PCT is not fully understood, but it is speculated that these drugs bind with porphyrins inside the lysosomes of liver cells, to be eventually excreted in the urine.
Hydroxychloroquine and chloroquine are contraindicated in pregnant women or women who are lactating. These drugs are also contraindicated for individuals with advanced liver disease, psoriasis, retinal disease, or glucose-6-phosphate dehydrogenase deficiency or who have recent or continued use of alcohol or drugs that are toxic to the liver (e.g. acetaminophen, isoniazid or valproic acid). Hydroxychloroquine and chloroquine can be associated with side effects including less serious ones (e.g., nausea, vomiting, headaches, etc.), but also more serious ones including seizures, muscle weakness or damage to the retinas of the eyes (retinopathy). Although retinopathy is unlikely with the low dose regimen used for PCT, an eye (ophthalmological) examination is recommended both before and after treatment. Signs of retinopathy can include blurred vision, light sensitivity or seeing halos around lights.
Iron chelators are drugs that bind to iron in the body allowing iron to be dissolved in water and excreted from the body through the kidneys. Iron chelators are less effective than phlebotomy or low dose hydroxychloroquine or chloroquine in treating individuals with PCT. However, these drugs may play a role in treating affected individuals in whom the use of the two front-line therapies is not possible, such as individuals with end stage renal disease who are on hemodialysis.
Affected individuals are advised to avoid environmental triggering factors of the disorder such as stopping alcohol consumption or smoking. The avoidance of sunlight may be necessary to protect the skin and can include the use of double layers of clothing, long sleeves, wide brimmed hats, gloves, and sunglasses. Pain killers (oral analgesics) can be used to treat painful skin lesion. Care should be taken to avoid infection of skin lesions. Antibiotics can be used to treat skin infections that do develop.
The treatment of PCT can achieve complete remission in affected individuals, but relapse is possible. The treatment of relapse is the same as the initial treatment.
Investigational Therapies
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office: Toll-free: (800) 411-1222 TTY: (866) 411-1010 Email: prpl@cc.nih.gov
For information about clinical trials sponsored by private sources, in the main, contact: www.centerwatch.com
The Porphyrias Consortium is a joint endeavor including five of the leading porphyria centers in the United States. Staff includes physicians, researchers, research coordinators, and technical laboratory staff. The Consortium aims to expand the knowledge about porphyrias to benefit patients and families. Study information regarding porphyrias is also posted at the Porphyrias Consortium website: http://rarediseasesnetwork.epi.usf.edu/porphyrias/index.htm
TEXTBOOKS Porphyria Cutanea Tarda. In: Handbook of Iron Overload Disorders. Barton JC, Edwards CQ, Phatak PD, et al. (eds). 2010 Cambridge University Press, New York, NY. Pp. 160-168.
Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of heme biosynthesis: X-linked sideroblastic anemias and the porphyrias. In: The Metabolic and Molecular Basis of Inherited Disease, 8th ed. Scriver CR, Beaudet AL, Sly WS, et al. (eds). 2001 McGraw-Hill, New York, NY. Pp. 2991.
Anderson KE. Porphyria Cutanea Tarda. In: NORD Guide to Rare Disorders. Lippincott Williams & Wilkins. Philadelphia, PA. 2003:493-494.
JOURNAL ARTICLES
Singal AK, Kormos-Hallberg C, Lee C, et al. Low-dose hydroxychloroquine is as effective as phlebotomy in treatment of patients with porphyria cutanea tarda. Clin Gastroenterol Hepatol. 2012;10:1402-1409. http://www.ncbi.nlm.nih.gov/pubmed/22985607
Frank J, Poblete-Gutierrez P. Porphyria cutanea tarda when skin meets liver. Best Pract Res Clin Gastroenterol. 2010;24:735-745. http://www.ncbi.nlm.nih.gov/pubmed/20955974
Phillips JD, Bergonia HA, Reilly CA, Franklin MR, Kushner JP. A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cutanea tarda. Proc Natl Acad Sci USA. 2007;104:5079-5084. http://www.ncbi.nlm.nih.gov/pubmed/17360334
Young LC. Porphyria cutanea tarda associated with Cys282Tyr mutation in HFE gene in hereditary hemochromatosis: a case report and review of the literature. Cutis. 2007;80:415-418.http://www.ncbi.nlm.nih.gov/pubmed/18189029
Sams H, Kiripolsky MG, Bhat L, Stricklin GP. Porphyria cutanea tarda, hepatitis C, alcoholism, and hemochromatosis: a case report and review of the literature. Cutis. 2004;73:188-190.http://www.ncbi.nlm.nih.gov/pubmed/15074347
Bygum A, Christiansen L, Petersen NE, et al. Familial and sporadic porphyria cutanea tarda: clinical, biochemical and genetic features with emphasis on iron status. Acta Derm Venereol. 2003;83:115-120. http://www.ncbi.nlm.nih.gov/pubmed/12735639
Egger NG, Goeger DE, Payne DA, et al. Porphyria cutanea tarda: multiplicity of risk factors including HFE mutations, hepatitis C, and inherited uroporphyrinogen decarboxylase deficiency. Dig Dis Sci. 2002;47:419-426. http://www.ncbi.nlm.nih.gov/pubmed/11855561
HEP is a deficiency of the enzyme uroporphyrinogen decarboxylase; it is the autosomal recessive form of f-PCT. The manifestations of HEP resemble Congenital Erythropoietic Porphyria (CEP), with symptoms of skin blistering that usually begin in infancy.
Skin photosensitivity results in severe blistering and scarring, often with mutilation and loss of facial features and fingers. Increased hair growth (hypertrichosis) on sun-exposed skin, brownish-colored teeth (erythrodontia), and reddish-colored urine are common. There may be bone fragility due to expansion of the bone marrow and vitamin deficiencies, especially vitamin D. Red blood cells have a shortened life-span, and mild or severe hemolytic anemia often results. Synthesis of heme and hemoglobin is actually increased to compensate for the shortened red blood cell survival and is associated with splenomegaly. Bacteria may infect the damaged skin and contribute to mutilation and scarring.
Who gets Hepatoerythropoietic Porphyria?
HEP is a very rare type of autosomal recessive porphyria. Each parent of an affected individual must have a mutation in one of their UROD genes and both must pass their mutation on to their child. This also means that both parents have f-PCT.
What causes Hepatoerythropoietic Porphyria?
HEP is caused by a deficiency of the enzyme uroporphyrinogen decarboxylase, due to the inheritance of mutations in both copies of a persons URO-decarboxylase genes.
How is Hepatoerythropoietic Porphyria diagnosed?
Diagnosis of HEP can be made by demonstrating significant elevations of specific porphyrins in urine and stool, as well as iidentification of a specific fluorescence emission peak in plasma. DNA testing to identify the specific mutations in an individuals UROD genes is the most specific and sensitive test to confirm the diagnosis of HEP.
What are treatments for Hepatoerythropoietic Porphyria?
Treatment is the same as for PCT: regularly scheduled phlebotomies (removal of blood) to lower the amount of porphyrins in the liver or a low dose regimen of hydroxychloroquine as well as removal of factors (for example, certain medications) that activated the disease and avoidance and/or protection from sunlight.
Additional Reading about HEP:
Hepatoerythropoietic Porphyria
NORD gratefully acknowledges Ashwani K Singal, MD, MSc, Division of Gastroenterology and Hepatology, University of Alabama at Birmingham, for assistance in the preparation of this report.
Synonyms of Hepatoerythropoietic Porphyria
autosomal recessive PCT
HEP
General Discussion
Summary
Hepatoerythropoietic porphyria (HEP) is an extremely rare genetic disorder characterized by deficiency of the enzyme, uroporphyrinogen decarboxylase. This deficiency is caused by mutations of both copies of a persons UROD gene, which means that the disorder is inherited as an autosomal recessive trait. Most affected individuals have a profound deficiency of this enzyme and onset of the disorder is usually during infancy or early childhood. However, some individuals may have a mild form that can go undiagnosed until adulthood. The childhood form of HEP is often associated with painful, blistering skin lesions that develop on sun-exposed skin (photosensitivity). Affected areas of skin can scar and become discolored. There may be risk of bacterial infection. Abnormal, excessive hair (hypertrichosis) on affected skin is also common. Mild anemia and abnormal enlargement of the liver and/or spleen (hepatosplenomegaly) have also been reported. Mild cases of HEP may go unrecognized until adulthood and can be clinically indistinguishable from porphyria cutanea tarda (PCT), a related disorder that may be acquired or occur in individuals with a mutation of one UROD gene (autosomal dominant inheritance). Cutaneous photosensitivity is generally much more severe in HEP than in PCT. NORD has a separate report on porphyria cutanea tarda.
Introduction
HEP belongs to a group of disorders known as the porphyrias. This group of at least seven disorders is characterized by abnormally high levels of porphyrins and porphyrin precursors due to deficiency of certain enzymes essential to the creation (synthesis) of heme, a part of hemoglobin and other hemoproteins. There are eight enzymes in the pathway for making heme and at least seven major forms of porphyria. The symptoms associated with the various forms of porphyria differ. It is important to note that people who have one type of porphyria do not develop any of the other types. Porphyrias are generally classified into two groups: the "hepatic" and "erythropoietic" types. Porphyrins and porphyrin precursors and related substances originate in excess amounts predominantly from the liver in the hepatic types and mostly from the bone marrow in the erythropoietic types. Porphyrias with skin manifestations are sometimes referred to as "cutaneous porphyrias". The term "acute porphyria" is used to describe porphyrias that can be associated with sudden attacks of pain and other neurological symptoms. HEP is a hepatic and cutaneous porphyria.
Signs & Symptoms
The symptoms and severity of HEP can vary from one person to another. Onset is usually within the first two years of life, but mild cases that go undiagnosed until adulthood have been reported. Although HEP is associated with specific, characteristic symptoms, several factors, including the small number of identified cases, make it difficult to establish the full range of associated symptoms of the disorder.
Severe cutaneous photosensitivity is usually the first sign. Affected infants may have extremely fragile skin that that can peel or blister on minimal impact is common. Reddening of the skin is common (erythema). Blistering skin lesions can develop on sun-exposed skin such as the hands and face. Photosensitivity can be severe and can cause scarring, erosion, and disfigurement. Bacterial infection of skin lesions can occur.
Abnormal, excessive hair growth (hypertrichosis) may also occur on sun-exposed skin. Affected skin may darken or lose color (hyper- or hypopigmentation). Small bumps with a distinct white head (milia) may also develop. Some affected individuals have teeth that are reddish-brown colored (erythrodontia).
Low levels of circulating red blood cells (anemia) may also occur. Anemia may be due to the premature destruction of red blood cells (hemolysis). Anemia associated with HEP may be mild or severe. Severe anemia may be associated with fatigue, pale skin, irregular heartbeat, chest pain, dizziness, and abnormally cold hands and feet. Some individuals may have an abnormally enlarged liver and/or spleen (hepatosplenomegaly).
Mild cases of HEP can go undiagnosed until adulthood. Overt photosensitivity may not be seen and mild skin damage can be mistaken for other conditions during childhood.
Causes
HEP is caused by mutations of both alleles of the UROD gene. Genes provide instructions for creating proteins that play a critical role in many functions of the body. When a mutation of a gene occurs, the protein product may be faulty, inefficient, or absent. Depending upon the functions of the particular protein, this can affect many organ systems of the body.
HEP is inherited as an autosomal recessive trait. Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Recessive genetic disorders occur when an individual inherits the same abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
Investigators have determined that the UROD gene is located on the short arm (p) of chromosome 1 (1p34.1). Chromosomes, which are present in the nucleus of human cells, carry the genetic information for each individual. Human body cells normally have 46 chromosomes. Pairs of human chromosomes are numbered from 1 through 22 and the sex chromosomes are designated X and Y. Males have one X and one Y chromosome and females have two X chromosomes. Each chromosome has a short arm designated p and a long arm designated q. Chromosomes are further sub-divided into many bands that are numbered. For example, chromosome 1p34.1 refers to band 34.1 on the short arm of chromosome 1. The numbered bands specify the location of the thousands of genes that are present on each chromosome.
The UROD gene creates (encodes) an enzyme known as uroporphyrinogen decarboxylase (UROD), which is the fifth enzyme in the heme biosynthetic pathway. In HEP, UROD enzyme activity is usually less than 10% its normal levels. Such low enzyme activity results in the abnormal accumulation of specific porphyrins and related chemicals in body, especially within the bone marrow, red blood cells, liver and skin. Symptoms develop because of this abnormal accumulation of porphyrins and related chemicals. For example when porphyrins accumulate in the skin, they absorb sunlight and enter an excited state (photoactivation). This abnormal activation results in the characteristic damage to the skin found in individuals with HEP. The liver removes porphyrins from the blood plasma and secretes it into the bile. When porphyrins accumulate in the liver, they can cause toxic damage to the liver.
Affected Populations
HEP is an extremely rare disorder that affects males and females in equal numbers. Approximately 40 cases have been reported in the medical literature. The exact incidence or prevalence of HEP in the general population is unknown.
Related Disorders
Symptoms of the following disorders can be similar to those of HEP. Comparisons may be useful for a differential diagnosis.
Congenital erythropoietic porphyria (CEP) is a rare inherited metabolic disorder resulting from the deficient function of the enzyme uroporphyrinogen III cosynthase (UROS), the fourth enzyme in the heme biosynthetic pathway. Due to the impaired function of this enzyme, excessive amounts of particular porphyrins accumulate, particularly in the bone marrow, plasma, red blood cells, urine, teeth, and bones. The major symptom of this disorder is hypersensitivity of the skin to sunlight and some types of artificial light, such as fluorescent lights (photosensitivity). After exposure to light, the photo-activated porphyrins in the skin cause bullae (blistering) and the fluid-filled sacs rupture, and the lesions often get infected. These infected lesions can lead to scarring, bone loss, and deformities. The hands, arms, and face are the most commonly affected areas. CEP is inherited as an autosomal recessive genetic disorder. Typically, there is no family history of the disease. Both parents are usually healthy, but each carries a defective gene that they can pass to their children. Affected offspring have two copies of the defective gene, one inherited from each parent. (For more information on this disorder, choose congenital erythropoietic porphyria as your search term in the Rare Disease Database.)
There are other conditions that may cause signs and symptoms that are similar to those seen in HEP. Such conditions include other cutaneous porphyrias, drug-induced photosensitivity, epidermolysis bullosa, various forms of lupus, and solar urticarial. (For more information on these disorders, choose the specific disorder name as your search term in the Rare Disease Database.)
Diagnosis
A diagnosis of HEP is based upon identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation and a variety of specialized tests. HEP may be considered in infants and children with chronic, blistering photosensitivity.
Clinical Testing and Workup
Screening tests can help diagnose HEP by measuring the levels of certain porphyrins in blood plasma, urine and red blood cells. These tests can help to differentiate the disorder from congenital erythropoietic porphyria by the different patterns of individual porphyrins and/or by demonstrating markedly decreased activity of the UROD enzyme. There is elevation of porphyrins in plasma, urine, and feces. Porphyrin patterns in HEP are similar to those seen in PCT with elevation of highly carboxylated porphyrins and isocoproporphyrins. In contrast to PCT, there are markedly increased levels of zinc protoporphyrin in red blood cells in HEP patients which is due to accumulation of pathway intermediates being metabolized to protoporphyrins.
Molecular genetic testing can confirm a diagnosis of HEP by detecting mutations in both UROD genes, but is available only on a clinical basis.
Standard Therapies
Treatment
The treatment of HEP is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, hematologists, dermatologists, hepatologists, and other healthcare professionals may need to systematically and comprehensively plan an affected childs treatment. Genetic counseling may benefit affected individuals and their families.
There is no specific, FDA-approved therapy for individuals with HEP. Because the disorder is so rare, most treatment information is based other forms of porphyria.
Avoidance of sunlight will benefit affected individuals and can include the use of clothing styles with long sleeves and pant legs, made with double layers of fabric or of light-exclusive fabrics, wide brimmed hats, gloves, and sunglasses. Topical sunscreens are generally ineffective, but certain tanning products with ingredients that increase pigmentation may be helpful. Affected individuals may also benefit from window tinting and the use of vinyl or films to cover the windows of their homes and cars.
Phlebotomies, which are used to treat individuals with PCT, are generally ineffective in individuals with HEP since elevated iron levels are not a feature of the disorder. Another treatment for PCT, the antimalarial drug chloroquine, was effective in at least one case reported in the medical literature.
Anemia may require treatment in some cases. Blood transfusions have been used to treat some individuals. Recombinant erythropoietin, which helps the body produce more red blood cells, was successfully used to treat severe anemia in an individual with HEP whose anemia was not associated with increased red cell destruction.
Investigational Therapies
Gene therapy is also being studied as another approach to therapy for individuals with genetic disorder associated with enzyme deficiency. In gene therapy, the defective gene present in a patient is replaced with a normal gene to enable the produce of the active enzyme and prevent the development and progression of the disease in question. Given the permanent transfer of the normal gene, which is able to produce active enzyme at all sites of disease, this form of therapy is theoretically most likely to lead to a cure. However, at this time, there remain some technical difficulties to resolve before gene therapy can be advocated as a viable alternative approach for genetic disorders like HEP.
To-Figueras J, Phillips JD, Gonzalez-Lopez JM, et al. Hepatoeythropoetic porphyria due to a novel mutation in the uroporphyrinogen decarboxylase gene. Br J Dermatol. 2011;165:499-505.http://www.ncbi.nlm.nih.gov/pubmed/21668429
Cantatore-Francis JL, Cohen J, Balwani M, et al. Hepatoerythropoietic porphyria misdiagnosed as child abuse: cutaneous, arthritic, and hematologic manifestations in siblings with a novel UROD mutation. Arch Dermatol. 2010;146:529-533. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3092549/
Armstrong DK, Sharpe PC, Chambers CR, et al. Hepatoerythropoietic porphyria: a missense mutation in the UROD gene is associated with mild disease and an unusual porphyrin excretion pattern. Br J Dermatol. 2004;151:920-923. http://www.ncbi.nlm.nih.gov/pubmed/15491440
Ged C, Ozalla D, Herrero C, et al. Description of a new mutation in hepatoerythropoietic porphyria and prenatal exclusion of a homozygous fetus. Arch Dermatol. 2002;138:957-960.http://www.ncbi.nlm.nih.gov/pubmed/12071824
Moran-Jimenez MJ, Ged C, Romana M, et al. Uroporphyrinogen decarboxylase: complete human gene sequence and molecular study of three families with hepatoerythropoietic porphyria. Am J Hum Genet. 1996;58:712-721. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1914669/
Tiffany England~ Stuck on you Vinyl~ Raises Awareness for APF rare disease!
Thursday - January 25, 2018 @ 06:00:43
Tiffany England~ Stuck on you Vinyl~ Raises Awareness for APF rare disease!
Order Here: https://www.facebook.com/stuckonyouvinyld/
Hi, My Name is Tiffany England. I was diagnosed with PCT and then VP at the age of 20. I've spent the last 16 years going from attack to attack, like most people do. I've had a couple years here and there where I didn't suffer from major attacks, but not often. Right now, is one of those good times.
So, about a year ago I started my own Vinyl Decal business. I make vinyl stickers that can go on your car, windows, walls, phone cases, signs, notebooks, computers, and more. It is something that I can do on my own time, when I'm feeling well. Its also something I really enjoy doing. Amy Chapman contacted me about a month ago to see if I would design one for the American Porphyria Foundation. I was so excited to do this. My hope is that they will find a cure or at least a better treatment for Porphyria in my lifetime. So, with saying this a portion of each sale will be donated to the American Porphyria Foundation.
The APF is not selling these. Please message me directly to place your order. Each item for sale is listed as A, B, C, D, E, F. They have the price and size listed on the picture. If you have questions please message me & I will respond promptly.
I wanted to specify that each decal is made custom and not going through a generic print/machine.
Thank you for stopping by my store, raising awareness about Porphyria! My hope is that you enjoy each one made personally.
Thanks, Tiffany England- Porphyria Advocate
What is ALAD & How common is it?
Monday - January 22, 2018 @ 17:14:18
ALAD-Deficiency Porphyria (ADP)
What is δ-Aminolevulinic Acid Dehydratase Porphyria?
ADP is a severe disorder caused by a deficiency of the enzyme δ-aminolevulinic acid dehydratase (ALAD) which results in an increase of 5-aminolevulinic acid (ALA) in the liver, other tissues, blood plasma, and urine. In addition, urine coproporphyrin and erythrocyte protoporphyrin are increased. ADP generally presents with sudden attacks of severe stomach pain that last for several days.
Who gets δ-Aminolevulinic Acid Dehydratase Porphyria?
All of the reported cases of ADP have been males, in contrast to the other acute porphyrias. ADP is the least common of all the porphyrias with less than 10 cases documented to date. This is an autosomal recessive disease, whereas the other three acute porphyrias are autosomal dominant. Each parent of an affected individual must have a mutation in one of their ALAD genes and both must pass their mutation on to their child.
What causes δ-Aminolevulinic Acid Dehydratase Porphyria?
ADP is caused by a deficiency of the enzyme δ-aminolevulinic acid dehydratase (ALAD).
How is δ-Aminolevulinic Acid Dehydratase Porphyria (ADP) diagnosed?
There are many laboratory tests available for the porphyrias, and it is often difficult to decide which should be chosen. Many of these tests are expensive and the results are often difficult to interpret. When abdominal and neurological symptoms suggest an acute porphyria, the best screening tests are urinary aminolevulinic acid (ALA) and porphobilinogen (PBG). DNA testing to identify the specific mutation in an individuals porphyria-causing gene is the most specific and sensitive test to confirm the diagnosis of a specific porphyria. Before requesting DNA testing, it is recommended that patients have biochemical testing (urinary, stool and/or plasma porphyrins and porphyrin precursors (ALA and PBG) and/or enzyme assays). However, biochemical testing may be inconclusive.
What are treatments for δ-Aminolevulinic Acid Dehydratase Porphyria?
Treatment is the same as in the other acute porphyrias. For the acute porphyrias, hospitalization is often necessary for acute attacks. Medications for pain, nausea and vomiting, and close observation are generally required with monitoring of salt and water balance. Harmful drugs should be stopped. Attacks are treated with either glucose loading or intravenous administration of hemin (Panhematin). Attacks can be prevented in many cases by avoiding harmful drugs and adverse dietary practices.
Miss Sian - Dancing through the shadows into the light
Monday - January 15, 2018 @ 06:00:48
The Story
Sian is a 12 year old, aspiring dancer from Topeka, KS. Her dedication, hard work, and determination has provided her a wonderful opportunity to participate in Motion 41's Summer Intensive Program in Omaha, NE. This opportunity would allow her to dance with older dancers from all over the US and learn from some amazing instructors from all over the world. Dancers strive for acceptance to a summer intensive program! Her total cost is $1,995.
Tuition $1,000. Room/meals $995. Most summer intensives cost $4000 for tuition/room/meals! She has a scholarship for $200, leaving a tuition balance of $800 due before May 18. I am saving money from each paycheck for the $995 room/board/meals. It will not be easy as I am a single parent. Si is working hard to earn money (baking and selling cookies, breads, pies; and doing chores). I'm afraid this may not be enough. Money raised will go to tuition, remaining room/meals, and incidentals. Let me share with you the story of Sian, and how you can be a part of her journey.
Dance - She was accepted into Ballet Midwest's Junior Company at the age of 10 and continues to be a member. She has performed in the Nutcracker as a solder, cadet and this year, en pointe, as a polichinette. She has performed roles in Swan Lake, Sleeping Beauty, and currently, Romeo and Juliet. When she is not in school, she is at the studio. Sometimes 7 days a week for 4 or more hours a day.
Why is this important to her? Dancing is her world! Dance allows her to feel and be normal. There are no boundaries, no limitations. She uses dance as a platform to talk about EPP, the FDA, and politics! She hides in the shadows every day of her life. Omaha is important to her because she can be normal, advance her skills, and dance out of the shadows into the light on a national level.
Why is this important to me? Si has every right to curl up in a ball and throw in the towel. She has every right to question why God has chosen her to carry these burdens. She has every right to be angry, bitter, jealous and envious. Instead, she pushes the raging inferno and injustice deep inside. She wipes away the tears, laces up the pointes, puts on a smile and performs. Everyone else sees a graceful, beautiful dancer with so much emotion and talent that will take her places. For her it's a moment that she is normal. Another wall broken down. Another chance to bring joy/happiness/inform people. For me, it's a moment to see a strong, determined young woman, who is fighting through the pain to change the world. I see perfection and confirmation as why God has chosen her. I see the daily pain that she is desperately trying to hide. I see the tears quickly wiped away so as others do not see. I hear the silent whys in her eyes, as she looks at me in physical or emotional pain from the name callers. Omaha is important to me, because it's her dream!
Medical - Si had all the odds stacked against her from birth. She was born 4 weeks premature, with a bicuspid aortic valve, an aortic stenosis, and a trivial aortic insufficiency. Most recently, she was diagnosed with NSHL (Neurosensori hearing loss) requiring a hearing aid and POTS (Postural Orthostatic Tachycardia Syndrome). All those conditions are nothing compared to the diagnosis that stopped us in our tracks -EPP. Her lip in the picture is from an EPP attack. Yet, she smiles through the torture.
Erythropoietic protoporphyria is a rare genetic blood disorder. Sian began to show symptoms of EPP when at age 4. These debilitating attacks are caused by exposure to any sunlight. Imagine being four, riding your bike in shorts, flip-flops, and not a care in the world. Abruptly, having to stay inside; wear long pants, long sleeves, hats, gloves, and sunglasses (year round). Not being able to swim, sled or play with friends outside! To be in constant pain from the light. Your skin and deep tissue burning like lava, stinging like a 1000 bees! There are no medications, pain relievers or treatments available to prevent, stop or assist. She sits in cold baths for hours, cold socks on her hands, fans blowing. Her attacks begin within 72 hours of exposure and can last at least 5 days. She has scars and pitting on her face and fingers. She endures rigorous blood tests every 6 weeks to check her CBCs, iron, TIBC, ferratin, hemoglobin, Vit D, iron and protoporphyrin levels. She endures feraheme infusions every 4 months at the Cancer Center due to EPP side effects. She is constantly being seen or in contact with her medical specialists at Children's Mercy (Hepatologist, Cardiologist, Otolaryngologist, Audiologist, and Dermatologist), specialists at the Cancer Center (Hematologist); Dr. Karl Anderson, specialist at UTMB; (Gastroenterologist); Dr. Joseph Bloomer, specialist at UAB (Gastroenterologist) and the APF.
APF (American Porphryia Foundation) According to the APF, "protoporphyrin accumulates first in the bone marrow. Then in red blood cells, plasma and sometimes the liver. Protoporphyrin is excreted by the liver into the bile, after which it enters the intestine and is excreted in the feces. It causes very painful photosensitivity and can greatly impair quality of life. Swelling, burning, itching, and redness of the skin may appear during or after exposure to sunlight, including sunlight that passes through window glass. This can cause mild to severe burning pain on sun-exposed areas of the skin. Skin manifestations generally begin early childhood and are more severe in the summer. There is an increased risk of gallstones, which contain protoporphyrin. Excess protoporphyrin can also cause liver damage. Less than 5% of EPP patients severe liver damage and a condition caused protoporphyric hepatopathy that sometimes requires liver transplantation."
Politics - There is no cure for this condition. The one medication that has proven to work is not available in the US! Clinuvel states on their website, "The FDA has recognised that afamelanotide meets an unmet clinical need and treats a severe genetic condition for patients who are life-long deprived of light. SCENESSE was granted orphan drug designation by the FDA in 2008 for the treatment of EPP patients and Fast Track Designation (FTD) in 2016, allowing for a rolling review of the NDA." Yet, is still unavailable for people who continue to hide from the sun. Sian is one of many EPP sufferers that have provided letters and pictures to the FDA in an attempt to show them what this condition is capable of doing and the impact it has on living a normal life.
Thank you for taking time to read! Even if you cannot make a donation, just the fact that we reached one more person and brought awareness to EPP, the APF, and Scenesse is rewarding.
If exercise were a pill, it would be the most widely prescribed medication in the world. (Emory University School of Medicine) Of all the things we can do for our health, few are more generally helpful than physical exercise.
Exert yourself. Leading a physically active life can help us feel happier, think more clearly, have more energy, be more productive and, along with proper diet, control our weight. Exercise need not be painful or extreme to be effective. Regular periods of moderate exercise several times a week can be very beneficial.
Jogging, brisk walking, biking, and taking part in active sportsenough both to get your heart beating faster and to cause you to break a sweatcan improve your endurance and help to prevent heart attack and stroke. Combining such aerobic exercise with moderate weight training and calisthenics helps to strengthen your bones, internal muscles, and limbs. These activities also contribute to maintaining a higher metabolism, which automatically helps to control your weight.
Exercise can be enjoyable
Use your feet. Exercise is beneficial for people of all ages, and membership in a gym is not required to get it. Simply using your feet instead of a car, bus, or elevator is a good start. Why wait for a ride when you can walk to your destination, perhaps even arriving there faster? Parents, encourage your children to participate in physical play, outdoors whenever possible. Such activity strengthens their bodies and helps them to develop whole-body coordination in ways that sedentary entertainment, such as video games, cannot.
No matter how old you are when you start, you can benefit from moderate physical exercise. If you are older or have health problems and have not been exercising, it is wise to consult a doctor about how to begin. But do begin! Exercise that is started gradually and not overdone can help even the oldest among us to maintain muscle strength and bone mass. It can also help seniors to avoid falls.
Exercise is what helped Rustam, mentioned in the first article of this series. Seven years ago, he and his wife began jogging a little each morning, five days a week. At first, we made excuses not to go, he relates. But having a partner helped to motivate both of us. Now it has become a good, enjoyable habit.
Patient Day In Orlando Florida ~ Its Free!
Tuesday - January 9, 2018 @ 18:15:20
Patient Day In Orlando Florida ~ Its Free! Complimentary Parking & A Full Day of learning about the Porphyrias, Meeting Experts & Q&A session Meet other patients and caregivers!
Saturday January 13, 2018 Patient Day 10:00 am Welcome D. Lyon-Howe, American Porphyria Foundation 10:05 am American Porphyria Foundation Overview Session 1- Heme Biosynthesis & the Erythropoietic Porphyrias Co-Chairs: E. Minder, MD & S. Keel, MD 10:15 am Heme Biosynthesis and the Porphyrias 101 J.D. Phillips, PhD 10:45 am Genetics 101 H. Naik, MS, CGC 11:00 am Erythropoietic Porphyrias Overview M. Balwani, MD, MS 11:30 am EPP Research J. Marcero 11:45 am Effective & Emerging Therapies for the Treatment of Erythropoietic Porphyrias E. Minder, MD, MS 12:15 pm Q & A moderated by Co-Chairs 12:45 pm Lunch Session 2- the Acute Hepatic Porphyrias Co-Chairs: D.M. Bissell, MD & C. Levy, MD 1:45 pm Diagnosis of the Porphyrias M. Badminton, MBChB, PhD 2:15 pm Acute Hepatic Porphyrias Overview D.M. Bissell, MD 2:45 pm Caregivers and Support Sandra Boone 2:45 pm Current & Emerging Treatments B. Wang, MD 3:30 pm Research Overview and Patient Perspective TBD and Sharon Dill, Patient Representative 3:45 pm Q & A moderated by Co-Chairs
4:30 pm Closing remarks American Porphyria Foundation
Hyatt Regency Orlando 9801 International Drive Orlando, Florida 32819 USA +1 (407) 2841234
Did you take part in a trial of Scenesse? We need your help!
NICE recently made a preliminary recommendation not make Scenesse available on the NHS in England. We are responding to their consultation document and need your help to make sure our response is as complete as it can be. Amongst other things, we are trying to demonstrate that the positive experiences of Afamelanotide are representative of most of those who were on the trial. But to do this we need to speak to people who were on any of the Scenesse trials to tell us how it affected them. Please get in contact on helpline@porphyria.org.uk or 0300 30 200 30. Or post a few sentences here.
THANK YOU for your continued support.
Wisdom for Heart and Health
Saturday - January 6, 2018 @ 06:00:26
Wisdom for Heart and Health
Wisdom for Heart and Health
A calm heart is the life of the fleshly organism.PROVERBS 14:30.
A heart that is joyful does good as a curer.PROVERBS 17:22.
Those simple but profound statements were spoken by King Solomon of Israel about three thousand years ago.* But are they true? What does modern medicine say?
Concerning a calm heart versus one given to anger, the Journal of the American College of Cardiology states: Current findings suggest a harmful association between anger and hostility and CHD [coronary heart disease]. Hence, the Journal notes: Successful prevention and treatment of CHD might involve . . . not only conventional physical and pharmacological therapies, but also psychological management focusing on anger and hostility. Simply put, a calm heart fosters good health, just as the Bible says.
Similar positive effects spring from a joyful heart. Dr. Derek Cox, a health official in Scotland, stated in a BBC News report: If you are happy you are likely in the future to have less in the way of physical illness than those who are unhappy. The same report stated: Happier people also have greater protection against things like heart disease and stroke.
What is more, that wisdom is recorded in plain language so that all can benefit. And it comes free of charge!
As millions have already discovered to their great joy, when wisdom enters into your heart and knowledge itself becomes pleasant to your very soul, thinking ability itself will keep guard over you, discernment itself will safeguard you.
Is that not most reassuring?
Shadow Jumpers Store Merchandise for Sale
Friday - January 5, 2018 @ 06:00:25
Did you know that we have "Shadow Jumpers"
Tshirts, Pullover Hoodies, New Wristbands, and hats
for Youth & Adults- Raise awareness today!
Here is the link and pricing gets yours today before they are gone`
Tiffany England ~ Stuck on you Vinyl~ Raising Awareness for Porphyria
Thursday - January 4, 2018 @ 13:14:07
Hi, My Name is Tiffany England. I was diagnosed with PCT and then VP at the age of 20. I've spent the last 16 years going from attack to attack, like most people do. I've had a couple years here and there where I didn't suffer from major attacks, but not often. Right now, is one of those good times.
So, about a year ago I started my own Vinyl Decal business. I make vinyl stickers that can go on your car, windows, walls, phone cases, signs, notebooks, computers, and more. It is something that I can do on my own time, when I'm feeling well. Its also something I really enjoy doing. Amy Chapman contacted me about a month ago to see if I would design one for the American Porphyria Foundation. I was so excited to do this. My hope is that they will find a cure or at least a better treatment for Porphyria in my lifetime. So, with saying this a portion of each sale will be donated to the American Porphyria Foundation.
Each listing can be viewed on FB at this link:
https://www.facebook.com/stuckonyouvinyld/
The APF is not selling these.
Please message me directly to place your order. Each item for sale is listed as A, B, C, D, E, F. They have the price and size listed on the picture. If you have questions please message me & I will respond promptly.
I wanted to specify that each decal is made custom and not going through a generic print/machine.
Thank you for stopping by my store, raising awareness about Porphyria! My hope is that you enjoy each one made personally.
Thanks,
Tiffany England- Porphyria Advocate
EXCITING ANNOUNCEMENT!! FREE Genetic Testing DNA
Thursday - January 4, 2018 @ 06:30:40
EXCITING ANNOUNCEMENT!!
You can now receive FREE genetic testing (DNA) and counseling for acute porphyria. DNA can tell a person if they carry a mutation in a gene associated with a predisposition for acute porphyria. This opportunity can shorten the time to diagnosis and prevent misdiagnosis.
How does it work?
*Genetic testing is ordered by your physician. *Results are then reported to your physician ONLY (No other organization will receive any identifiable information regarding a patient or their results)
Who is eligible?
*Patients must be 16 to 55 years old *Patients must meet eligibility from at least one of the following criteria:
Elevated Urinary Porphobilinogen (PBG)
OR
Unexplained recurrent (more than one), prolonged (>24 hours) episodes of severe, diffuse abdominal pain AND at least 2 of the following:
Red to brownish urine, OR Known or suspected family history of an acute hepatic porphyria, OR Blistering skin lesions on sun-exposed areas, OR Peripheral nervous system manifestations occurring around the time of abdominal pain (i.e. motor neuropathy (paresis), sensory neuropathy (numbness, tingling, limb pain), OR Central nervous system manifestations occurring around the time of abdominal pain (i.e. confusion, anxiety, seizures, hallucinations), OR Autonomic nervous system manifestations occurring around the time of abdominal pain (i.e. hyponatremia, tachycardia, hypertention, nausea and vomiting, constipation) This program was developed to reduce barriers to genetic testing and counseling and to help people make more informed decisions about their health.
Contact the APF on 1-866-APF-3635 or 713-APF-3635 to guide you through the process.
Patient Day In Orlando Florida ~ Its Free!
Thursday - January 4, 2018 @ 06:00:02
Patient Day In Orlando Florida ~ Its Free! Complimentary Parking & A Full Day of learning about the Porphyrias, Meeting Experts & Q&A session Meet other patients and caregivers!
Saturday January 13, 2018 Patient Day 10:00 am Welcome D. Lyon-Howe, American Porphyria Foundation 10:05 am American Porphyria Foundation Overview Session 1- Heme Biosynthesis & the Erythropoietic Porphyrias Co-Chairs: E. Minder, MD & S. Keel, MD 10:15 am Heme Biosynthesis and the Porphyrias 101 J.D. Phillips, PhD 10:45 am Genetics 101 H. Naik, MS, CGC 11:00 am Erythropoietic Porphyrias Overview M. Balwani, MD, MS 11:30 am EPP Research J. Marcero 11:45 am Effective & Emerging Therapies for the Treatment of Erythropoietic Porphyrias E. Minder, MD, MS 12:15 pm Q & A moderated by Co-Chairs 12:45 pm Lunch Session 2- the Acute Hepatic Porphyrias Co-Chairs: D.M. Bissell, MD & C. Levy, MD 1:45 pm Diagnosis of the Porphyrias M. Badminton, MBChB, PhD 2:15 pm Acute Hepatic Porphyrias Overview D.M. Bissell, MD 2:45 pm Caregivers and Support Sandra Boone 2:45 pm Current & Emerging Treatments B. Wang, MD 3:30 pm Research Overview and Patient Perspective TBD and Sharon Dill, Patient Representative 3:45 pm Q & A moderated by Co-Chairs
4:30 pm Closing remarks American Porphyria Foundation
Hyatt Regency Orlando 9801 International Drive Orlando, Florida 32819 USA +1 (407) 2841234
Jessi Brill and battling with EPP
Wednesday - January 3, 2018 @ 06:00:43
JESSI BRILL lives in Northern Virginia. I was diagnosed with EPP about 5 years ago, but my first episode was when I was 18 months old. As, I'm sure, many of you can relate, I ran the gauntlet growing up trying to figure out what was wrong with me. After being told that it was anything from having inactive sweat glands that caused swelling to just flat out being told it was all in my head, I stopped going to see doctors until I was 25 when I found the APF. It was after my parents were watching "Mystery Diagnosis" and saw the preview of the next episode of the little boy who was having reactions to the sun. They looked at each other after only watching the preview and said "That's Jess".
After making the 7 hr. drive to Charlotte, NC I met with Dr. Bonkovsky who tested me and all the years of no answers narrowed down to a 1 hr. meeting with him telling me that Im not crazy and there is something causing this pain and these episodes. I only have my reactions in the warmer months. I have never had a reaction during the winter. And although I love the winter, I have never been skiing or snowboarding in my life so no extended trips outside. Dr. Bonkovsky thinks that this is the reason I've never had a reaction during the winter months.
I have a 1 year old little girl (Lexi) that I pray doesn't have to go through what I did my entire life. I remember all too well what it was like growing up being the kid who couldn't go out and play. I don't want that for her. Even though what I know now would make things completely different for her, still.
So, my question for anyone willing to share for those of you with EPP who have kids, how do you do it? We went on our first family vacation this past summer and of course it was to the beach. I lasted 2 days with limited time in the sun before I couldn't do it anymore. The rest of my family was there so they took her out for me but it doesn't stop the emotions or tears from getting overwhelming when you know you can't do stuff with your child because of this. Editor's note:Porphyrias are family diseases, which affect every member of the family.
A 32-year-old woman presented to the emergency department (ED) with 3 months of abdominal pain and 1 week of vomiting.
The differential diagnosis of abdominal pain is broad. This presentation could be caused by disorders of the gastrointestinal (GI), gynecologic, urinary, or, less likely, the neuromuscular systems. The presence of vomiting supports a GI cause. Pregnancy should be excluded in any woman of childbearing age presenting with abdominal pain.
Characteristics of the pain, including location, temporal characteristics, severity, and aggravating and alleviating factors, can narrow the differential diagnosis. The past medical history, including prior surgeries, menstrual, and obstetric history, is also critical.
Approximately 3 months prior to presentation, she reported a tick bite that had evolved into a circumferential targetoid rash. Her primary care provider performed serologic testing for Lyme disease, which was negative, and prescribed doxycycline, which she stopped after a week because of nausea and diffuse, achy, and constant abdominal pain. After initial improvement, symptoms recurred a week prior to presentation. The nausea was now associated with intractable vomiting and anorexia. She denied hematemesis or coffee ground emesis. Her abdominal pain intensified and radiated to her back. She lost 10 pounds over the past week. She denied headache, constipation, diarrhea, blood per rectum, melena, dysuria, vaginal discharge, or rash. She reported chills and temperatures up to 37.8 ° C at home.
She had a history of migraine headaches for which she took ibuprofen occasionally but took no other prescription or over-the-counter medications. She had never smoked, consumed 2 alcoholic beverages a month, and denied illicit drug use. She lived with her boyfriend on a farm in Indiana where she raised chickens, rabbits, and ducks.
The patient dates the onset of nausea and abdominal pain to a course of doxycycline, presumably prescribed for early Lyme disease, which was stopped after only 1 week. GI side effects, including nausea, vomiting, and upper abdominal pain, are common with doxycycline and may account for the early symptoms. However, these symptoms typically resolve promptly with drug discontinuation. Doxycycline may rarely cause esophageal and gastric ulcers, which could explain her symptoms.
Fewer than half of patients with erythema migrans caused by Lyme disease are seropositive at presentation, as there has been insufficient time for antibodies to develop. Lyme disease typically affects the skin, joints, heart, and nervous system and only rarely affects the GI tract. Acute Lyme disease can cause intestinal pseudoobstruction, splenomegaly, and mild hepatitis. Although Lyme disease is unlikely to be the cause of the current symptoms, serologic testing should be repeated and should be positive if the patient now has early disseminated disease.
Patients with Lyme disease are occasionally coinfected witha second organism. Ixodes scapularis, the tick that transmits Lyme disease in the Northeast and Midwest, can be coinfected with Babesia microti, a red cell parasite. Babesiosis can persist for months and presents with fever, malaise, and many other nonspecific symptoms, including some that this patient has: anorexia, weight loss, abdominal pain, and vomiting.
The history of migraine and intractable vomiting suggests the possibility of cyclic vomiting syndrome. This syndrome is characterized by episodic bouts of vomiting lasting from hours to as long as a week. The vomiting is often accompanied by abdominal pain and occasionally headaches. Episodes are separated by asymptomatic periods that may last months. Cyclic vomiting syndrome can occur at any age but is more common in children, those with a personal or family history of migraines, and heavy users of cannabis. At least 3 stereotypical episodes are required to make the diagnosis, so a history of prior similar symptoms should be explored.
The differential diagnosis of abdominal pain and vomiting should stay broad until a comprehensive physical exam and initial laboratory tests are performed. Volume status should be assessed by estimating jugular venous pressure and by obtaining supine and standing blood pressure measurements. The abdomen should be examined carefully, and the presence or absence of hepatomegaly, splenomegaly, masses, and ascites should be specifically noted. The presence of bradycardia, oligoarticular arthritis, or neuropathy could provide supporting evidence for Lyme disease. Pregnancy is less likely given the diffuse and persistent nature of the pain but should still be excluded.
On physical examination, she was distressed, writhing on the bed, and appearing comfortable only on her side with her knees flexed. Her temperature was 36.5 ° C, heart rate 83 beats per minute, respiratory rate 18 breaths per minute, blood pressure 143/77 mmHg, and oxygen saturation 94% while breathing ambient air. Her abdomen was diffusely tender, most markedly in the epigastrium. Abdominal rigidity, rebound tenderness, and costovertebral tenderness were absent. There was no rash; the previously reported targetoid skin lesion was no longer present. The remainder of the exam was normal.
Laboratory evaluation showed a white count of 7900/mm3, hemoglobin 14.3 gm/dL with normocytic indices, and a platelet count of 175,000/mm3. Sodium was 130 mmol/L, potassium was 3.1 mmol/L, bicarbonate 26 mmol/L, blood urea nitrogen 15 mg/dL, creatinine 0.6 mg/dL, and glucose 92 mg/dL. Serum calcium, aspartate aminotransferase, alanine aminotransferase, bilirubin, and lipase were normal. A urine pregnancy test was negative. Urine analysis was negative for nitrites and leukocyte esterase. Abdominal and pelvic computed tomography (CT) scan with intravenous (IV) contrast performed 3 days prior at an outside ED revealed a 3.4 centimeter left ovarian cyst. A subsequent transvaginal ultrasound was negative for cyst torsion and confirmed appropriate placement of an intrauterine device.
The absence of abdominal rigidity and rebound tenderness does not exclude peritonitis. A normal white blood cell count also does not reliably exclude serious intraabdominal pathology. However, the CT scan argues strongly against many common causes of abdominal pain, including appendicitis, diverticulitis, perforated ulcer, intestinal obstruction, and malignancy, assuming the symptoms have not changed since it was performed.
The patients laboratory studies argue against biliary obstruction, pancreatitis, pregnancy, hypercalcemia, and ongoing urinary tract infection. Patients with functional gallbladder disorders may have normal laboratory and CT findings but typically have recurrent, biliary-colic-type pain. The low serum potassium, a high blood urea nitrogen to creatinine ratio, and a low serum sodium reflect her significant vomiting. The hyponatremia is consistent with the appropriate release of antidiuretic hormone (ADH) in the setting of volume depletion. She should receive isotonic fluids plus potassium in addition to symptomatic treatment of pain and nausea. Given the severity and duration of symptoms, an esophagogastroduodenoscopy (EGD) should be performed to exclude GI mucosal disease, including peptic ulcer disease and gastritis, which may not be evident on the CT scan.
Additional diagnoses should be considered at this point. This patient has exposure to chickens, ducks, rabbits, and ticks as well as reported chills and mild temperature elevation at home. Tularemia, which can be transmitted by tick bites or exposure to infected rabbits, can cause a prolonged illness. Some patients have abdominal pain, anorexia, nausea, and weight loss, although fever is usually more prominent. Tularemia is uncommon and most frequently seen in the south-central part of the United States but has been reported throughout the country. She should be queried regarding additional exposures, including well water to assess her risk for Campylobacter infection.
Opiate withdrawal can present with pain and vomiting, but she reports no opiate use and lacks other findings such pupillary dilation or piloerection. Given the prevalence of opiate abuse, however, a toxicology screen should be performed. Hypercalcemia and diabetic ketoacidosis as metabolic causes of abdominal pain have been ruled out by her laboratory values. If no other cause is identified, other metabolic etiologies like Addison disease, familial Mediterranean fever, or porphyria should be considered.
Cyclic vomiting syndrome should still be on the differential. It is a diagnosis of exclusion requiring a history of recurrent, stereotypical episodes, which should be explicitly explored.
The patient was admitted to a medical unit by the hospitalist service and received IV normal saline, parenteral potassium, and IV pantoprazole. She underwent an EGD that revealed minor erosions in the antrum of the stomach. Biopsies were obtained.
Seven hours after the endoscopy, the patient had a brief period of confusion followed by a generalized tonic-clonic seizure lasting 1 minute. A head CT without contrast was negative for any focal abnormality. Repeat laboratory evaluation revealed that serum sodium was 125 mmol/L, and serum glucose was 113 mg/dL. She was transferred to the progressive care unit and received IV levetiracetam.
The endoscopy excluded structural abnormalities of the stomach and duodenum. The patient now has an additional problem, seizure, which needs to be incorporated in the diagnostic reasoning.
Seizures can be caused by the rapid development of severe hyponatremia, with serum sodium levels usually less than 120 mmol/L. Seizures caused by hyponatremia are typically preceded by headache and lethargy, as the intracellular movement of excess water causes cerebral edema. Hyponatremia is unlikely to be the cause of her seizure but should nevertheless be evaluated with a urine sodium concentration and serum and urine osmolality. If she is euvolemic, the IV fluids should be stopped and her free water intake should be restricted to avoid worsening the hyponatremia, as it is potentially caused by the syndrome of inappropriate ADH (SIADH).
There are many other possible causes for new onset seizures in adults, including brain tumor, head trauma, alcohol withdrawal, medications, and central nervous system infection, including Lyme disease. Lyme serologies should be repeated.
In this patient, it is likely that the seizure is a manifestation of the same illness that is causing her vomiting and abdominal pain. Seizure is not a feature of cyclic vomiting syndrome in adults. It is also not a feature of tularemia, adrenal insufficiency, or opioid withdrawal.
Acute intermittent porphyria (AIP) can cause both abdominal and neurologic problems. Hyponatremia is common during acute attacks, caused by either the inappropriate release of ADH or the appropriate release of the hormone if there is fluid loss. AIP is a rare diagnosis but could explain the uncommon combination of abdominal pain, vomiting, seizure, and hyponatremia. A spot urine porphobilinogen test should be sent to assess for AIP.
Additional laboratory studies were sent. Serum osmolality was 269 mosm/kg with a corresponding urine osmolality of 699 mosm/kg. A random urine sodium was 145 mEq/L. Thyroid stimulating hormone and cosyntropin stimulating testing were normal. IgM and IgG antibodies to Borrelia burgdorferi were negative. Urine porphobilinogen was sent. An electroencephalogram did not reveal epileptiform discharges. Magnetic resonance imaging (MRI) of the brain was significant for T2/FLAIR hyperintensity in the cortex and subcortical white matter of the occipital lobes bilaterally. Hypertonic saline and fluid restriction were initiated.
The patients labs are consistent with SIADH. Excessive ADH release because of volume depletion and consequent hyponatremia should have improved rapidly with the administration of saline. The high urine sodium suggests that she is now volume replete, while the high urine osmolality is consistent with the presence of excessive ADH in the absence of appropriate stimuli. In the context of normal thyroid and adrenal function, the hyponatremia is likely due to the SIADH.
Negative serologic testing for Lyme disease, 3 months after the onset of rash, excludes this diagnosis.
The MRI findings are consistent with posterior reversible encephalopathy syndrome (PRES), a clinicoradiographic syndrome of headache, altered mental status, seizure, and/or vision loss with associated white matter abnormalities of the posterior cerebral hemispheres. PRES has been reported with AIP as well as other disorders, most commonly hypertensive encephalopathy, eclampsia, and immunosuppressive drug use.
The patients sodium improved with fluid restriction and the administration of hypertonic saline. There was no recurrence of seizure activity. Amlodipine was initiated for blood pressure readings as high as 156/106 mmHg. A hepatobiliary scan revealed a gallbladder ejection fraction of 13%. Biopsies from her endoscopy revealed nonspecific inflammation without the presence of Helicobacterpylori. The patient was discharged home 7 days after admission after stabilization of serum sodium, improvement in her abdominal pain, and tolerance of oral intake. A plan was made for outpatient cholecystectomy.
Many causes of abdominal pain have been excluded and the remaining diagnostic possibility, porphyria, is rare. The clinicians have revisited their differential and considered other causes of abdominal pain, including functional gallbladder disorders. However, chronic cholecystitis (or functional gallbladder disorder) is not this patients primary problem. The diffuse, severe, and constant abdominal pain prior to admission is not typical of biliary pain, and many medical conditions and drugs, including amlodipine, can lead to a positive hepatobiliary scan. Chronic cholecystitis would not explain her seizure.
AIP remains at the top of the differential for this young woman. A urine porphobilinogen has been sent and must be followed up prior to any further workup or surgery.
One week after discharge, the patients urine porphobilinogen resulted at 172.8 mCmol/ (upper limits of normal 8.8). Sequencing analysis for genes coding the enzymes involved in the synthetic pathway for heme were sent. Hydroxymethylbilane synthase, coproporphyrinogen oxidase, and protoporphyrinogen oxidase mutation assays were all normal. Despite the normal genetic assays, the diagnosis of AIP was made on the basis of the clinical presentation and elevated urine porphobilinogen. The patient was referred to a hematologist and initiated on oral glucose supplements and hematin infusions.
DISCUSSION
Although abdominal pain has a broad differential, the combination of abdominal pain and neurologic or psychiatric symptoms should suggest the possibility of porphyria, especially if symptoms are recurrent or unexplained. The porphyrias are a group of disorders caused by defects in the synthetic pathway of heme, leading to an overproduction and accumulation of precursors. Heme is a component of multiple proteins, including hemoglobin, myoglobin, and the cytochrome P450 enzymes. Although it is synthesized in all tissues, the bone marrow and liver are the organs most actively involved. The porphyrias can be classified according to the primary site of the overproduction and accumulation of heme precursors (liver vs bone marrow). Although there is overlap between the 2 groups, hepatic porphyrias often present with acute neurovisceral symptoms, while the erythropoietic porphyrias often cause cutaneous photosensitivity.1
AIP is the most common hepatic porphyria with a prevalence of 1 in 20,000 in Caucasians of Western European descent.1 AIP is caused by a defect in the gene that encodes porphobilinogen deaminase, leading to the accumulation of porphobilinogen.1The cardinal manifestation is an acute porphyric attack. While the precise mechanisms underlying the symptoms are unknown, the accumulating metabolites may be directly neurotoxic.2 Attacks are precipitated by factors that induce heme synthesis, including caloric restriction, alcohol, and certain medications, particularly those that upregulate cyP450. The most commonly implicated drugs are anesthetics, antiepileptics, sulfonamides, rifampin, and estrogen and progesterone. Attacks can also be precipitated by changes in endogenous sex hormone levels, like the increase in progesterone seen in the luteal phase of the menstrual cycle, which may account for the higher incidence of symptomatic attacks in women.3
Acute attacks of AIP may have a wide variety of presentations; the disease was referred to as the little imitator in the early 20th century.4 The most common symptom is acute, severe abdominal pain, which may mimic an acute abdomen. Because the pain is neuropathic rather than inflammatory, abdominal tenderness, rebound, fever, and leukocytosis are usually absent, as they were in this patient. Abdominal pain is often accompanied by neuropsychiatric symptoms, including sensory and motor neuropathy, anxiety, hallucinations, delirium, and altered level of consciousness. Seizure occurs in 20% of cases. Involvement of the autonomic nervous system causes tachycardia and new onset hypertension in the majority of patients as well as restlessness and tremor. Hyponatremia, mediated by the syndrome of inappropriate ADH secretion, occurs in nearly a third of patients.5,6 MRI findings consistent with PRES have also been described in AIP.7
The diagnosis of AIP is often delayed; diagnosis later in the disease course is associated with a poorer prognosis.8 Reported intervals between presentation and diagnosis range from several months to as long as 20 years.9 Associating the use of medications, caloric restriction, or the menstrual cycle with the exacerbation of symptoms or darkening of urine can help prompt an earlier diagnosis.6
AIP can be diagnosed by detecting a greater than 5-fold elevation of urinary porphobilinogen excretion in conjunction with the typical symptoms of an acute attack.5 Renal dysfunction causes urinary excretion of PBG to fall and serum levels to rise.10 Serum PBG levels should therefore be sent when AIP is suspected in the setting of renal dysfunction. The primary role of genetic testing in a patient who has AIP confirmed clinically and biochemically is to assist in genetic counseling and to identify asymptomatic family members.11 Genetic testing is not required to confirm the diagnosis and does not help prognosticate. It is unusual that a mutation was not detected in this case, as the current sensitivity of genetic testing is 97% to 100%.11
There are 4 principles of management of an acute porphyric attack. First, any precipitating factors such as medications should be stopped. Second, abdominal pain should be treated appropriately with opioids, if necessary. Third, if autonomic dysfunction is present, beta-blockers or clonidine should be given to treat hypertension.5 Finally, glucose and/or hemin should be administered to downregulate aminolevulinic acid (ALA) synthase by negative feedback. Downregulation of ALA synthase decreases the accumulation of the neurotoxic porphyrin precursors ALA and PBG.5 For patients with mild symptoms, glucose alone (300-500 g/d) may be enough to abort the attack.12 This can be achieved via a high-carbohydrate diet in those able to tolerate oral intake or via continuous infusions of dextrose containing fluids.5 For more severe attacks with associated polyneuropathy, respiratory muscle weakness, or seizures, or for attacks that are not resolving, heme preparations dosed at 3 to 4 mg/kg/d for 3 to 4 days are indicated.5
The recent diagnosis of acute Lyme disease was a distractor in this presentation. In Lyme endemic areas, patients with erythema migrans are treated based on the clinical presentation rather than serologic testing.13 Although this patient took only 1 week of doxycycline, testing during this hospitalization showed that she had either been cured early or had not had Lyme disease in the first place. There is no known association between Lyme disease and the porphyrias, and doxycycline is not a common precipitant of AIP attacks.14 However, the GI side effects of doxycycline may have decreased caloric intake and ultimately provoked the patients first attack of AIP. The clinicians in this case appropriately avoided the target but hit the mark by correctly diagnosing AIP.
KEY POINTS
Consider AIP in patients with unexplained abdominal pain, especially when accompanied by neuropsychiatric symptoms and autonomic lability.
Diagnose AIP by sending a urine PBG during a suspected acute attack.
Treat AIP acutely by removing precipitants, treating abdominal pain, and initiating dextrose-containing fluids and hemin infusions to downregulate ALA synthase.
Acknowledgments
The authors thank the patient who enthusiastically supported the writing of this report.
Disclosure
Warren Gavin, MD has disclosed participation in expert testimony. The authors have no financial or other conflicts of interest to disclose.
2018 Goals Can you and you family do some of these?
Monday - January 1, 2018 @ 05:00:00
Being a better person is the top New Years resolution, according to a new poll from Marist College in New York.
Their findings showed that about 12 percent of those surveyed want to become a better person in 2018. The same amount reported wanting to lose weight in the new year. Only 7 percent of those surveyed want to improve their overall health, and 9 percent would like to eat healthier, exercise more or get a better job.
While New Years resolutions are a big focus this time of year, the survey revealed that only about 44 percent of Americans were planning on creating goals for 2018. For those who do want to make changes but arent sure where to start, here are 15 ideas of easy, but meaningful, changes one can make:
Eat more fish. As we previously reported, a recent study indicated that fish helped children sleep better and improved IQs by up to four points when eaten every week. Omega-3 fatty acids found in fish also have been linked to lower rates of heart disease.
Make meaningful connections. Health professionals at the Aspen Ideas Festivalrecently spoke about how an increasing amount of people feel lonely, which takes a toll on mental and physical health. While you can have many friends and still feel lonely, talking about your feelings with others can provide social support.
Take a warm bath. According to a story on Bravo, a study from March indicated that a hot bath of 104 degrees could burn as many calories as a 30-minute walk.
Go to bed early. Poor sleep is linked to diabetes, heart disease, obesity and depression, according to the Centers for Disease Control. Create a self-imposed bedtime, set an alarm as a reminder, and aim to hit the hay in time to get a solid seven to eight hours.
Volunteer. According to an article published by Harvard Health Publications, giving your time can lower blood pressure, and even helps those who volunteer frequently live longer.
Meal prep. And no, this doesnt mean you cant enjoy a meal out. Research from Johns Hopkins University showed that people who make their own meals consume fewer calories than those who dont cook.
Give up soda (yes, even diet). While some opt to drink calorie-free beverages for weight-loss purposes, a recent study indicated that people who regularly consumed diet soft drinks were at an increased risk for dementia or stroke, reported the The Washington Post.
Hit the gym. Aside from all the obvious reasonsweight management and heart healthresearch has shown that exercising is good for your brain, too. A study published last month indicated that physical activity could improve memory and slow aging.
Dont eat after 9 p.m. Yes, youve heard this many times before, but as we previously reported, late-night snacking goes against our biological clocks. A study from earlier this year indicated that eating later in the evening could increase your heart disease risk.
Pick up a book. Reading is good for your brain, but health professionals say that the hobby also can enrich your social skills. People who read fiction may understand people better than others," cognitive psychologist Keith Oatley explained to CNN. "A work of fiction is a piece of consciousness that can pass from one mind to another and that reader can make it their own."
Eat salad once a week. In a story published earlier this month, Newsweekreported that eating leafy greens weekly could keep your brain up to 11 years younger.
Spend more time outside. Mounting research shows that enjoying nature and green space can help lower stress and may reduce symptoms of depression and anxiety.
Get creative. Remember the adult coloring book craze? Well, they have benefits other than serving as a way to fight boredom. "Coloring definitely has therapeutic potential to reduce anxiety, create focus or bring [about] more mindfulness," certified art therapist Marygrace Berberian told CNN in a story last year.
Use social media less. Its good to be connected, but even Facebook recently admitted that too much time online could be bad for your mental health. Researchhas shown that children and teens in particular may have lower self esteem due to social medias influence. Limit how much time you spend each day and replace that online FOMO with quality time spent networking IRL.
Save for the future. Its easy to get caught up in the day-to-day expenses, but saving for your financial future is important. As CNN reported, many Americans dont even have $500 stored away for emergencies. Skip happy hour once a month, buy generic or utilize the library for books, and immediately transfer the money you saved into a separate account.
Important Research Notice for Families.
Wednesday - October 10, 2018 @ 12:22:30
Important Research Notice for Families.
You and your family are needed for a very important research project. We need at least 25 families. Even if you are already on a study, you can be added to this research project, too.
The parameters are as follows:
You must be a woman with recurrent or cyclical attacks of an acute porphyria (AIP, VP or HCP).
Both of your parents need to be alive or one parent should be alive and diagnosed with an acute porphyria.
One of your siblings must be diagnosed and have attacks. Your other siblings are also needed on the study. They do not need to be diagnosed or have symptoms.
Please contact Desiree at the APF if you are interested in participating in this important research. 866. APF.3635 or lyonapf@aol.com
"Remember..Research is the key to your cure!"
It has been known for just over 50 years that a sm
Monday - February 19, 2018 @ 09:12:31
It has been known for just over 50 years that a small dose (15 mg) of zinc sulfate will stop an AIP attack in minutes, at a cost of pennies per dose, with no hospitalization and no side effects. Why won't APF endorse simple cost effective Zinc Sulfate therapy for AIP?
Dr. Suzy Melkonian has been providing high quality
Wednesday - March 21, 2018 @ 19:36:10
Dr. Suzy Melkonian has been providing high quality medical services in Hematology and Oncology in the Mission Hills, Porter Ranch, and the Greater San Fernando Valley for over 20 years. Hematology
hello im bob bishop and i cant get the doctors to
Friday - April 27, 2018 @ 17:52:38
hello im bob bishop and i cant get the doctors to test my genetics! im in seattle washinton I have all the symptoms and have been to the cancer care allance here but they said you dont have blisters where you should and assured me that a 24 hour urine would show it !
Thank you for sharing this about caffeine and suga
Monday - May 28, 2018 @ 05:17:00
Thank you for sharing this about caffeine and sugar habits. I also have AIP and cannot tolerate much caffeine as it really effects my neuropathy. As far as watching our intake on the sugary sweets, this was a challenge for me at first. Especially when my Doctor suggested that I eat a whole chocolate cake to avoid an attack when symptoms started. Of course, I knew not to do that however the idea to snack on foods with sugar does seem to be pretty mainstream. I have consulted with Nutritionists over the years and have learned healthy choices that give the same benefits without the fear of weight gain and diabetes. I would encourage anyone with this disease to take this into consideration for your long term health.
Granuloma Annulare is a skin condition or type of
Sunday - June 3, 2018 @ 20:13:03
Granuloma Annulare is a skin condition or type of rash. Granuloma annulare may causes no symptoms but Granuloma Annulare Natural Treatment have been reported to help at least some cases making diagnosis relatively easy.
it was to late. Would have never had the chance
Saturday - June 9, 2018 @ 06:21:16
it was to late. Would have never had the chance to see 5 beautiful grands or the chance to raise my son. What is the entourage effect?
Granuloma Annulare is a skin condition or type of
Thursday - June 28, 2018 @ 03:56:13
Granuloma Annulare is a skin condition or type of rash. Granuloma annulare may causes no symptoms but Granuloma Annulare Natural Treatment have been reported to help at least some cases making diagnosis relatively easy.
Granuloma Annulare Natural Treatment
Monday - July 23, 2018 @ 04:23:21
Granuloma Annulare Natural Treatment information about the Symptoms, Causes and Diagnosis. Natural Herbal Treatment for Granuloma Annulare with Herbal Product Greneton Natural Supplement for chronic skin condition.
Thank you for this nice blog,its very informative.
Wednesday - July 25, 2018 @ 09:09:43
Thank you for this nice blog,its very informative.- Service Animal trained to perform a specific task for their handler's disability.Service Dogs have full access to all public places including restaurants, hotels, and no-pet housing. A fee cannot be a charge for granting access to Service Dogs. A service dog is considered a medical device and not a pet.Emotional Support Animal
Nice blog post This blog clearly shows the impo
Thursday - July 26, 2018 @ 09:32:45
Nice blog post This blog clearly shows the importance of genetic testing. With the help of at home genetic testing you can do genetic testing.
Thanks for sharing valuable information about DNA
Monday - August 6, 2018 @ 09:28:50
Thanks for sharing valuable information about DNA testing for porphyria. DNA test is really helpful to detect future health. Original Gene
useful information on topics that plenty are inter
Monday - August 27, 2018 @ 16:44:35
useful information on topics that plenty are interested on for this wonderful post.Admiring the time and effort you put into your b!.. Sleep disorders
Hey guys! this really helped me so i decided to le
Tuesday - August 28, 2018 @ 03:04:16
Hey guys! this really helped me so i decided to let you all know about this because it's gonna help you too regardless of your diseases or virus you are sick of. I was cure of Parkinson's disease of 4 years by a herbal medicine from MADIDA HERBAL CENTER, their herbal medicine is really good with zero side effect and it works awesomely. according to them on their websit: www.madidaherbalcenter.weebly.com they have cures to different diseases and virus like HIV, HERPES, CANCER, LUPUS, ASTHMA, HEPATITES and many more. Email them now @ madidaherbalcenter@gmail.com to place your order to get well and with collect effort & crucial information like this we can win the battle of ill health.<<<>>>><<>>><<>>> I feels really great now. ////////
Sian is an amazing little girl, and you are a bles
Tuesday - August 28, 2018 @ 05:20:40
Sian is an amazing little girl, and you are a blessing of a mother to her. Keep up the good work, Kim.
Granuloma Annulare Natural Treatment information a
Wednesday - September 5, 2018 @ 06:30:31
Granuloma Annulare Natural Treatment information about the Symptoms, Causes and Diagnosis. Natural Herbal Treatment for Granuloma Annularee with Herbal Product Greneton Natural Supplement for chronic skin condition.
Polycythemia Vera Natural Treatment Learn about th
Tuesday - November 27, 2018 @ 20:32:36
Polycythemia Vera Natural Treatment Learn about the Symptoms, Causes and Diagnosis. Natural Herbal Treatment of Polycythemia by Herbal Product Veraceton is a Natural Supplement for stem cell disorder.
An adjustment in eating regimen and way of life wi
Tuesday - December 11, 2018 @ 03:42:48
An adjustment in eating regimen and way of life with subsequent decrease in corpulence can likewise particularly lessen the seriousness of indications. sleep with valium
Interesting post and amazing tips. I really like t
Friday - December 14, 2018 @ 10:28:49
Interesting post and amazing tips. I really like this post. Thank you so much for sharing this post. For more info visit here: Mita Burke
Thank you for sharing your journey.
Friday - December 14, 2018 @ 15:42:57
Thank you for sharing your journey.
Feel free to find me on facebook if you have any q
Saturday - December 15, 2018 @ 01:22:14
Feel free to find me on facebook if you have any questions. You can message me anytime. Just search Victor LaFae. - Cheers




 Welcome to Shadow Jumpers! This is a page for EPP kids and parents by EPP kids and parents. Shadow Jumpers was created to help give kids with Erythropoietic Protoporphyria (EPP) and their families a place to learn about this rare disease, read tips and tricks learned over time and to hear from fellow kids. Through spreading awareness, fellow EPP interviews, tips to protecting yourself outside and some insight for parents, we hope all families living with EPP will look at this condition as a challenge they can overcome. Be sure to check out all the Shadow Jumper resources! Wed love to hear from you. Reach out to us anytime on shadowjumpers@porphyriafoundation.org!!
Welcome to Shadow Jumpers! This is a page for EPP kids and parents by EPP kids and parents. Shadow Jumpers was created to help give kids with Erythropoietic Protoporphyria (EPP) and their families a place to learn about this rare disease, read tips and tricks learned over time and to hear from fellow kids. Through spreading awareness, fellow EPP interviews, tips to protecting yourself outside and some insight for parents, we hope all families living with EPP will look at this condition as a challenge they can overcome. Be sure to check out all the Shadow Jumper resources! Wed love to hear from you. Reach out to us anytime on shadowjumpers@porphyriafoundation.org!!





















 The disorders Acute Intermittent Porphyria (AIP), Hereditary Coproporphyria (HCP), Variegate Porphyria (VP) and ALAD Porphyria (ADP) are treated initially with the administration of carbohydrate/glucose. This therapy has its basis in the ability of glucose to decrease porphyrin biosynthesis in the liver.
The disorders Acute Intermittent Porphyria (AIP), Hereditary Coproporphyria (HCP), Variegate Porphyria (VP) and ALAD Porphyria (ADP) are treated initially with the administration of carbohydrate/glucose. This therapy has its basis in the ability of glucose to decrease porphyrin biosynthesis in the liver.











































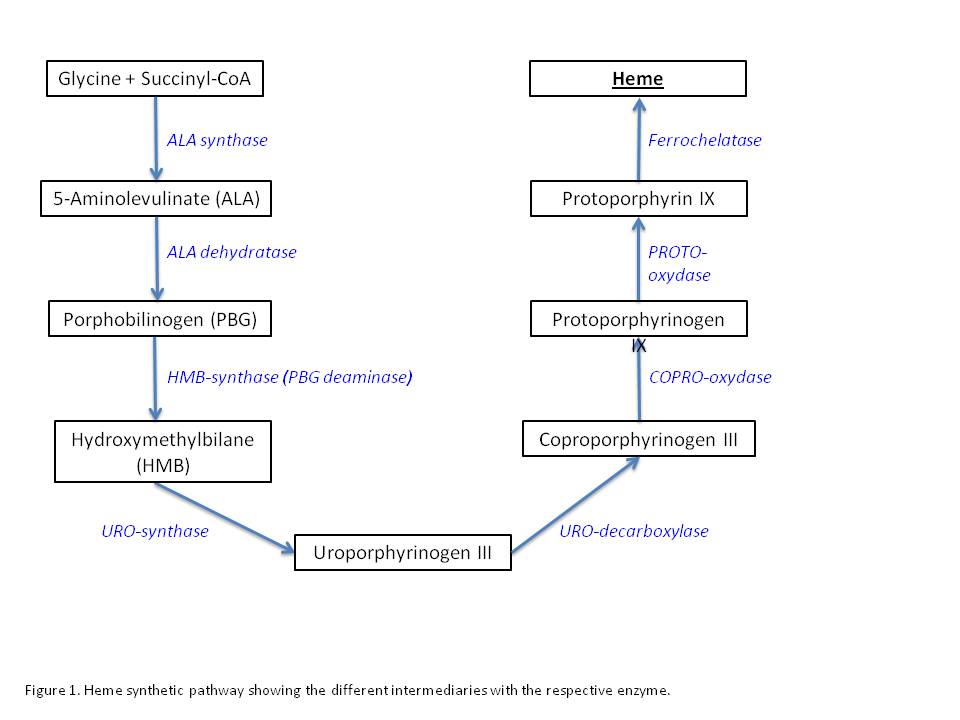










































 t in their life. Despite this, there is no cure for the majority of rare diseases and many go undiagnosed. Rare Disease Day improves knowledge amongst the general public of rare diseases while encouraging researchers and decision makers to address the needs of those living with rare diseases.
t in their life. Despite this, there is no cure for the majority of rare diseases and many go undiagnosed. Rare Disease Day improves knowledge amongst the general public of rare diseases while encouraging researchers and decision makers to address the needs of those living with rare diseases.
 Since Rare Disease Day was first launched by
Since Rare Disease Day was first launched by 

 The first Rare Disease Day was celebrated in 2008 on 29 February, a rare date that happens only once every four years. Ever since then, Rare Disease Day has taken place on the last day of February, a month known for having a rare number of days.
The first Rare Disease Day was celebrated in 2008 on 29 February, a rare date that happens only once every four years. Ever since then, Rare Disease Day has taken place on the last day of February, a month known for having a rare number of days.































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}